Hiperparatireoidismo secundário: uma complicação da Doença Renal Crônica
Secondary hyperparathyroidism: a Chronic Kidney Disease complication
Rafael A. Porto1
Marília R. Truite2
Sérgio Elias Gardano Bucharles3
Aline Borsato Hauser4
1Farmacêutico(a) (UP) atuante em análises clínicas em Curitiba, PR, Brasil.
2Médico nefrologista do Instituto do Rim do Paraná com doutorado pela PUC-PR – Curitiba, PR, Brasil.
3Professora do Curso de Farmácia da Universidade Federal do Paraná – UFPR – Curitiba, PR, Brasil.
Artigo recebido em 16/05/2013
Artigo aprovado em 29/01/2016
Resumo
A Doença Renal Crônica (DRC) é considerada um problema de saúde pública mundial e sua incidência vem aumentando progressivamente, chegando a ser classificada como uma epidemia. Uma de suas complicações é a Doença Mineral Óssea, uma desordem sistêmica que envolve alterações clínicas, bioquímicas e ósseas. Uma das causas desse distúrbio é o hiperparatireoidismo secundário, complicação decorrente da alteração do metabolismo de cálcio e fósforo, que representa uma importante causa da perda da qualidade de vida dos pacientes. Na DRC ocorre um quadro de hiperfosfatemia e hipocalcemia devido à perda da função renal, ao déficit de vitamina D ativa, e pelo desequilíbrio na manutenção do produto cálcio-fósforo. Como consequência à hipocalcemia ocorre um aumento na secreção do paratormônio, responsável pela reabsorção de sais ósseos, elevando os níveis de Ca+ no líquido extracelular. A tendência resultante é o estabelecimento de um quadro de HPTS, que pode causar fraturas patológicas, deformidades ósseas e decréscimo na sobrevida dos pacientes. Esta revisão sugere o HPTS como alvo de futuras investigações clínicas e laboratoriais para melhor elucidar os mecanismos envolvidos no desenvolvimento desta complicação e, assim, estabelecer o uso de medidas terapêuticas e profiláticas que aumentem a expectativa de vida dos pacientes com DRC.
Palavras-chave
Doença renal crônica; Hiperparatireoidismo secundário; Fatores de risco
INTRODUÇÃO
A National Kidney Foundation (NKF) define a Doença Renal Crônica (DRC) como uma lesão presente por um período igual ou superior a três meses, definida por anormalidades estruturais ou funcionais dos rins, manifestada por alterações patológicas que resultam na presença de biomarcadores de lesão renal em análises de soro ou urina.(1)
Na DRC ocorre uma perda progressiva e irreversível da função renal, o que resulta em uma redução da taxa de filtração glomerular (TFG) e acúmulo de toxinas urêmicas. Esta situação leva ao desenvolvimento, em estágios avançados da doença, da chamada síndrome urêmica, com manifestações que se assemelham a uma intoxicação sistêmica causada por uma ou várias substâncias dialisáveis.(2)
Atualmente, a DRC é considerada um problema de saúde pública mundial e, portanto, devem ser aplicadas políticas de saúde no Brasil e no mundo para minimizar esta situação.(3) Além de apresentar alta morbidade e mortalidade, sua incidência vem aumentando progressivamente segundo Censo de 2011 da Sociedade Brasileira de Nefrologia (SBN) (Figura 1), chegando a ser classificada como uma doença epidêmica de alto custo.(4) Em 2002, estimava-se terem sido gastos R$ 1,4 bilhão no tratamento de pacientes em diálise crônica e com transplante renal.(5)
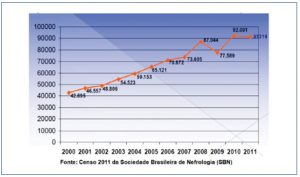
Figura 1. Total estimado de pacientes em tratamento dialítico por ano.
As doenças renais levam a óbito aproximadamente 15 mil pessoas por ano no país, sendo usualmente silenciosa no início. O diagnóstico precoce evita que ocorra falência renal, além de reduzir as chances de complicações.(6,7)
Levando-se em conta os fatores de risco tradicionais (Tabela 1) para o desenvolvimento da DRC, o aumento da prevalência de diabetes e a hipertensão arterial sistêmica (HAS) contribuem significativamente para o aumento do número de pacientes com DRC.(8)

A DRC é dividida em cinco estágios funcionais, de acordo com o grau de função renal do paciente, sendo que, quando a TFG cair para <15 mL/min é necessário submeter o paciente à diálise ou transplante renal (Tabela 2). A presença da DRC pode ser detectada por meio de teste para detecção de proteinúria e dosagem de creatinina sérica para avaliar a TFG. O reconhecimento e o manejo precoces de pacientes em estágios iniciais da DRC podem reduzir o aumento no número dos pacientes urêmicos.(3-5,9)

Em sua evolução, a DRC é acompanhada por um desequilíbrio hidroeletrolítico, metabólico e do sistema imunológico, visto que o processo inflamatório crônico é comprovado pela presença de níveis mais elevados de citocinas inflamatórias neste grupo de pacientes, apesar dos mecanismos envolvidos não estarem bem elucidados.(10) Assim, a doença é caracterizada pela perda das funções bioquímicas e fisiológicas de todos os sistemas do organismo, secundária ao acúmulo de toxinas urêmicas, surgindo alterações como hiponatremia, hipercalemia, acidose metabólica, hiperfosfatemia, hipocalcemia e hipervolemia, e as complicações associadas como anemia, aterosclerose acelerada, calcificação vascular, resistência à insulina, aumento do catabolismo muscular, perda de apetite, aumento da permeabilidade da membrana peritoneal e doença mineral óssea (DMO), que representam importantes indicadores de mortalidade por doença cardiovascular (DCV) e doenças infecciosas neste grupo de pacientes.(2,10,11) A HAS também é uma manifestação da DRC, caracterizada pelo volume circulante excessivo, podendo ser controlada com redução da ingestão de sódio, diuréticos potentes e remoção do líquido pela diálise. O excesso de volume circulante pode desencadear o aparecimento de insuficiência cardíaca congestiva (ICC) e edema agudo de pulmão.(2,12)
O trabalho em questão aborda o hiperparatireoidismo secundário (HPTS), uma complicação decorrente da alteração do metabolismo de cálcio e de fósforo em pacientes com DRC, uma das formas de apresentação da DMO. Esta complicação determina um estado de alto turnover ósseo, caracterizado por uma alta atividade osteoblástica e osteoclástica.(13) A importância do estudo do HPTS se encontra no impacto negativo na qualidade de vida e na alta morbimortalidade destes pacientes, uma vez que essa doença contribui diretamente para o aumento de fraturas ósseas, hospitalização e mortalidade, devido à redução da densidade mineral óssea.(14)
A PRODUÇÃO DE PTH E O METABOLISMO DE CÁLCIO E FÓSFORO
As glândulas paratireoides normalmente apresentam-se na forma de quatro nódulos esféricos de coloração rósea, localizadas na parte posterior da tireoide.(15) São responsáveis pela produção do PTH, que é metabolizado pelo fígado e excretado pelos rins.(16-18) A principal função do PTH é estimular a liberação de cálcio para o plasma, e, para isso, age principalmente nos ossos, nos rins e indiretamente no intestino delgado.(19)
Nos ossos, o PTH estimula a reabsorção óssea permitindo o acoplamento entre osteoblastos e osteoclastos, aumentando de uma forma geral o turnover ósseo. Doses intermitentes de PTH associam-se a um predomínio da formação, enquanto que exposição prolongada e concentrações elevadas induzem a uma perda óssea por predomínio da reabsorção.(17) Nos rins, o PTH age diretamente nos túbulos renais, aumentando a reabsorção de cálcio e diminuindo a reabsorção de fósforo e de bicarbonato.(19) Como ação indireta no intestino delgado, o PTH estimula, via 1-a-hidroxilase, a biossíntese renal de 1,25 (OH)2D3, a qual age promovendo a absorção intestinal de cálcio e fosfato da dieta, com consequente feedback negativo nas glândulas paratireoides.(17)
A matriz óssea é composta por uma parte orgânica (35%) e uma parte inorgânica (65%), que constitui a parte mineral hidroxiapatita. O cálcio e o fósforo são os principais constituintes minerais do esqueleto, uma vez que ele contém 98% de cálcio e 80% do fósforo do organismo. O cálcio da dieta é absorvido por transporte ativo no intestino, principalmente no duodeno e jejuno. Esse transporte ocorre na borda-em-escova das células epiteliais e envolve uma ATPase dependente de cálcio e uma proteína ligadora de cálcio, chamada de calbindina D.(20) A síntese dessa proteína e o funcionamento do transporte ativo dependem da vitamina D ativa.(21)
Aproximadamente 99% do cálcio do organismo estão armazenados nos ossos, sendo apenas 1% no líquido extracelular e 0,1% no líquido intracelular. Portanto, o osso atua como grande reservatório para o armazenamento de cálcio e como fonte de rápida mobilização quando a concentração desse íon no líquido extracelular ocasionalmente diminui. Quando a concentração de cálcio do líquido extracelular cai abaixo da normal, as glândulas paratireoides são diretamente estimuladas, promovendo a produção e secreção de PTH. A seguir, este hormônio atua diretamente sobre os ossos, aumentando a reabsorção de sais ósseos, liberando grandes quantidades de cálcio no líquido extracelular. Quando a concentração de íons cálcio se apresenta elevada, a secreção de PTH diminui, de modo que não ocorre quase nenhuma reabsorção óssea. Outro fator que influencia a reabsorção de cálcio é a concentração plasmática de fosfato para manter o produto cálcio e fósforo em níveis circulantes adequados.(22)
A vitamina D apresenta duas vias de formação: a via endógena e a via exógena. A vitamina D exógena apresenta-se sob duas formas: D2 (ergocalciferol), sintetizada em plantas a partir do precursor ergosterol e a D3 (colecalciferol), dos alimentos não vegetais. Ambas sofrem o mesmo processo de metabolização para se tornarem ativas. A vitamina D3 é produzida a partir do 7-diidrocolesterol (pró-vitamina D3), precursor imediato do colesterol. Por ação da radiação solar (ultravioleta B), é transformada em pré-vitamina D3, que sofre uma isomerização induzida pelo calor e forma vitamina D3. Esta atinge a circulação, sendo transportada até o fígado, onde se inicia o processo de hidroxilação.(23) A vitamina D é convertida no fígado na sua forma intermediária (25(OH)D), a qual é o principal metabólito circulante da vitamina D. As concentrações séricas de 25(OH)D refletem tanto a entrada quanto a produção endógena e pode ser medida para se verificar o nível geral de vitamina D. No rim, a forma 25(OH)D sofre uma segunda hidroxilação e é convertida pela enzima 1-a-hidroxilase em sua forma ativa (1,25(OH)2D3), a qual possui um importante papel na integridade dos ossos e músculos por regular o metabolismo do cálcio. O nível sérico da vitamina D na sua forma ativa não é relacionado com o estado geral de vitamina D e, por isso, não é clinicamente útil.(24)
O HIPERPARATIREOIDISMO SECUNDÁRIO NA DOENÇA RENAL CRÔNICA
O HPTS na DRC é caracterizado pela hiperplasia das glândulas paratireoides, sendo uma complicação frequente nos pacientes em diálise, podendo estar presente mesmo em fases da DRC, porém sendo mais prevalente nas fases avançadas da doença. Vários são os fatores responsáveis pela patogênese da doença, destacando-se a hiperfosfatemia, a hipocalcemia e o déficit de vitamina D.(25)
No paciente com DRC, o balanço de fósforo é alterado devido à perda dos néfrons, desta forma há uma redução nas taxas de excreção de fósforo, levando à hiperfosfatemia.(4) A retenção de fósforo pode também funcionar como inibidor indireto da produção da forma ativa da vitamina D devido à inibição da enzima 1-a-hidroxilase renal, que é responsável pela conversão da vitamina D em sua forma ativa (1,25(OH)2D3). Como consequência da diminuição da vitamina D ativa, ocorre uma redução na absorção intestinal e reabsorção óssea de cálcio, favorecendo o desenvolvimento de episódios mais frequentes e sustentados de hipocalcemia.(25)
Assim, a perda progressiva de massa renal determina queda dos níveis circulantes da forma ativa da vitamina D e uma drástica redução na absorção intestinal de cálcio. A tendência resultante à hipocalcemia leva ao estabelecimento de um quadro de HPTS, que permite manter o cálcio plasmático em nível normal ou pouco reduzido à custa de uma mobilização das reservas ósseas e do estabelecimento de um balanço negativo de cálcio, resultando em uma progressiva descalcificação óssea, uma vez que o PTH vai buscar no reservatório ósseo o cálcio que deveria provir da absorção intestinal.(2)
Para agravar ainda mais a situação, pacientes dependentes de diálise crônica apresentam grande retenção de fósforo aumentando o produto cálcio e fósforo acima do nível crítico, gerando na maioria das vezes calcificações ectópicas, principalmente vasculares. Este processo pode ter consequências graves como a obstrução coronariana e a complicação por DCV.(2,17)
O HPTS é considerado uma das mais importantes causas de mortalidade em pacientes com DRC por causar fraturas, deformidades ósseas e decréscimo do tempo de vida desses pacientes.(26) Em estudos epidemiológicos feitos com grande número de pacientes portadores de DRC, observa-se que até 80% dos mesmo apresentam valores de PTH acima dos valores laboratoriais normais de referência, especialmente aqueles que se encontram em programa regular de terapia renal substitutiva (hemodiálise e diálise peritoneal).(27) Analisando apenas o grupo composto por pacientes em tratamento dialítico, aproximadamente 50% podem desenvolver esta complicação em formas mais graves da doença e a mesma se associar a outras complicações, como desnutrição, também muito prevalente na população em tratamento dialítico.(28)
Um estudo com base na dosagem de PTH sérico revelou uma incidência de 56% de HPTS em pacientes com DRC em fase terminal. Tal estudo demonstrou uma direta correlação entre tempo de diálise e o aparecimento da doença, uma vez que, após o segundo ano de tratamento dialítico, a incidência do HPTS chegava a 70% destes pacientes.(29) Estudos demonstram que, no Brasil, a prevalência das lesões ósseas decorrentes do HPTS elevou-se de 32,3% na década de 80 para 44% nos anos 90.(30)
A principal característica dessa doença é a hiperplasia das glândulas paratireoides, que leva ao aumento da síntese e secreção do PTH(31) devido à diminuição da concentração sérica de cálcio, que leva ao decréscimo na síntese de calcitriol e à retenção de fósforo, além da resistência esquelética à ação do PTH.(31) Esta situação atenua a excreção de cálcio na urina e mobiliza o cálcio dos ossos, mantendo as concentrações séricas normais, ou seja, preservando a homeostase deste cátion.(32) (Figura 2).
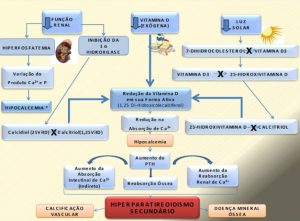
Figura 2. Representação esquemática da fisiopatologia do hiperparatireoidismo secundário na DRC.
Fonte: os autores
A hipocalcemia é um poderoso estimulante para a secreção de PTH e para o crescimento das glândulas da paratireoide. Os efeitos do cálcio parecem ser mediados pelo receptor de cálcio, e vários estudos têm demonstrado que existe um decréscimo da expressão do receptor de cálcio nas glândulas hiperplásicas que são vistas nos doentes renais. O decréscimo dos receptores de cálcio pode levar potencialmente a um aumento nos níveis de secreção de PTH porque a resposta da glândula da paratireoide frente ao estímulo do cálcio pode estar diminuída. Entretanto, a relação entre a expressão do receptor de cálcio e os níveis de PTH não é clara.(33)
Apesar da osteodistrofia renal (complicações do osteometabolismo associadas à Doença Renal Crônica) ser conhecida há mais de 50 anos, os conceitos sobre os mecanismos envolvidos nesta doença sofreram modificações e evoluíram com o tempo.(34) Atualmente, a DMO é considerada uma desordem sistêmica manifestada por uma ou mais combinações de anormalidades,(35) envolvendo alterações clínicas, bioquímicas e ósseas. Dentre as alterações bioquímicas, destacam-se às ocorridas nos metabolismos de cálcio, fósforo, PTH e vitamina D, e, em relação às alterações ósseas, destacam-se as anormalidades no turnover, mineralização, volume ou crescimento ósseo, além das calcificações extraósseas como calcificação vascular ou de outros tecidos.(14,36) Esta situação, antes conhecida como osteodistrofia renal (ODR), atualmente foi conceituada pelo KDIGO como DMO. Assim, o termo ODR ficou reservado para as alterações na histologia óssea avaliadas por biópsia e DMO corresponde a uma situação mais ampla desta complicação.(37) Estas anormalidades estão inter-relacionadas e provavelmente irão variar de acordo com o tipo particular e a rapidez da progressão da DRC, bem como em função de características individuais dos pacientes.(33)
Pacientes nos primeiros estágios da DRC raramente apresentam evidências clínicas de DMO, porém, estudos recentes têm revelado que alterações histológicas podem se desenvolver antes do aparecimento dos sintomas clínicos da doença.(38) A DMO pode ser classificada conforme o estado de turnover ósseo. As formas de alto turnover têm um perfil histológico definido como osteíte fibrosa, incluídas também as formas leves. A única causa desse distúrbio é o HPTS. No outro extremo se identificam as formas de baixo turnover, que, em função da taxa de mineralização, se dividem em: osteomalácia e enfermidade óssea adinâmica.(32,39-41) Muitos dos pacientes com DRC apresentam os dois tipos da doença óssea, sendo que a acidose metabólica pode intensificar este processo.(38,39)
A DMO em pacientes com doença renal muitas vezes é assintomática, e os sintomas aparecem somente nos estágios finais. Muitos deles são inespecíficos(36) e incluem dores ósseas, fraturas, deformidades ósseas, miopatia, ruptura de tendão e retardo de crescimento. Em adição, estas alterações metabólicas e suas terapias prolongadas podem provocar calcificações de tecidos.(42) Como a atividade aumentada dos osteoclastos e dos osteoblastos e a extrema velocidade de remodelação dificultam a mineralização adequada dos novos ossos, como consequência tem-se um osso estruturalmente fraco e com baixa densidade.(43) A osteosclerose é uma manifestação relacionada à atividade excessiva dos osteoclastos em resposta a reabsorção óssea, ou pela produção aumentada de osteoides mineralizados.(44) As lesões ósseas que aparecem podem variar muito entre os pacientes, devido a fatores de idade, etiologia da doença, dieta, tratamento instituído e duração do tratamento dialítico.(14) A remodelação óssea desencadeia ou acelera, também, a calcificação vascular, a qual é um risco para o desenvolvimento de DCV.(37) A calcificação vascular é extremamente comum e importante nos pacientes com doença renal, nos quais se desenvolve e progride rapidamente e prediz uma variedade de consequências adversas.(33) Na DRC, qualquer forma histológica da doença óssea renal pode estar associada aos níveis altos ou baixos de densidade óssea; assim, a densidade mineral óssea e os critérios da OMS não podem ser usados para o diagnóstico de osteoporose neste grupo de pacientes.(45)
A maioria dos pacientes com HPTS em fase terminal não responde ao tratamento clínico e, após alguns anos de diálise, removem as glândulas da paratireoide (paratireoidectomia total) quando o HPTS não responde mais ao tratamento farmacológico.(46) Todas as glândulas são removidas, mesmo as glândulas menores e macroscopicamente normais, que podem ser responsáveis pela persistência e retorno da doença no futuro.(28)
Para o diagnóstico do HPTS em pacientes com DRC são realizados testes laboratoriais de marcadores do metabolismo ósseo, incluindo dosagem de PTH, cálcio e fósforo.(33,47) A dosagem de fósforo e cálcio deve ser feita frequentemente e a terapia precisa ser ajustada de acordo com o guia de prática clínica para manter as concentrações dentro dos limites definidos.(33) Os marcadores bioquímicos têm sido utilizados há muito tempo para análise do desenvolvimento da DMO no paciente com DRC. A dosagem de PTH é usada amplamente por ser o hormônio regulador do turnover e da atividade celular óssea. Outros marcadores que refletem a formação óssea estão sendo avaliados, como: fosfatase alcalina total, fosfatase alcalina osso-específica, pró-colágeno tipo I, peptídeo terminal-C e osteocalcina. Por outro lado, encontram-se os marcadores da reabsorção óssea, como: fosfatase ácida tartrato resistente tipo 5b (TRAP-5b), colágeno telopeptídeo tipo1 (ICTP) e osteoprotogerina (OPG). Estes marcadores podem fornecer informações do turnover ósseo, mas nenhum garante suficiente correlação com o estado clínico da DMO.(48)
A maioria dos estudos de perda óssea na DRC depende da absormetria radiológica de dupla energia (DXA), a qual mede a densidade mineral óssea.(49) Porém, testes de densidade óssea não distinguem entre os diferentes tipos de histologia do osso.(50) A biópsia óssea é um método gold standard para determinar o estado ósseo por meio de estudos histológicos, entretanto, é um método invasivo que requer tempo de análise. Assim, é de grande interesse o desenvolvimento de técnicas de imagem não invasivas que possam melhorar a previsão de risco de fraturas em pacientes com DRC.(48,51,52)
O projeto das diretrizes do Kidney Disease Quality Initiative (K/DOQI) foi desenvolvido e tem sido divulgado amplamente pela NKF no intuito de que sejam adotadas práticas de diagnóstico, prevenção e tratamento das doenças renais baseadas nas melhores evidências dos trabalhos científicos. Dentre essas, algumas são específicas para a abordagem das alterações do metabolismo mineral. De acordo com estas recomendações, pacientes em diálise devem manter as concentrações de cálcio sérico corrigido para a albumina entre 8,4-9,5 mg/dL, fósforo sérico entre 3,5-5,5 mg/dL, produto cálcio x fósforo menor que 55 mg/dL e PTH plasmático entre 150-300 pg/mL. O tratamento do HPTS continua sendo um desafio para a área médica. Ele deve incluir uma combinação da restrição de fósforo na dieta, análogos da vitamina D, uso de calcimiméticos e de quelantes de fósforo. Assim, o primeiro passo é aperfeiçoar os níveis de fósforo e cálcio séricos. Isto pode ser alcançado por meio de dieta de restrição e pelo início da terapia com quelantes de fósforos. O segundo passo deve ser focado no controle dos níveis de PTH e vitamina D pelo uso de calcimiméticos (Cinacalcete) e/ou análogos da vitamina D. Por último, o controle de doses de quelantes de fósforo, calcimiméticos e análogos da vitamina D devem ser ajustados para se alcançarem os valores recomendados pela NKF.(53-56)
CONSIDERAÇÕES FINAIS
A DRC é considerada uma epidemia mundial, onde o alto custo do tratamento é uma preocupação para os órgãos governamentais e, por isso, um diagnóstico precoce da doença é fundamental. O HPTS é uma complicação da DRC e, por alterar o metabolismo de cálcio e fósforo, pode acarretar a DMO. O aumento do PTH, a hiperfosfatemia, a hipocalcemia e a deficiência de vitamina D ativa são os principais fatores da gênese do HPTS, podendo levar a calcificação vascular, dores e deformidades ósseas.
O HPTS, por ser uma importante causa de mortalidade e de perda da qualidade de vida em pacientes com DRC, deve ser alvo de estudos adicionais para que o paciente possa receber o tratamento ideal aumentando sua expectativa de vida. Assim, esta revisão sugere futuras investigações em estudos clínicos para elucidar os mecanismos envolvidos no HPTS a DRC, assim como o uso de medidas terapêuticas e de profilaxia desta complicação.
Abstract
Chronic Kidney Disease (CKD) is considered a global public health problem and its incidence has been progressively increasing, nearly becoming an epidemic. One of its complications is the Mineral Bone Disease, which is a systemic disorder that involves clinical, biochemical or bone alteration. One of its causes is the secondary hyperparathyroidism, a complication resulting from alterations of the metabolism of calcium and phosphorus and results a decrease in quality of life in patients. The CKD causes hyperphosphatemia and hypocalcemia due to kidney damage, active vitamin D deficiency and maintenance of calcium – phosphorus product. As a result of the hypocalcemia, the secretion of the parathyroid hormone increases, which is responsible for the bone reabsorption, increasing the levels of calcium in the extracellular liquid. The resultant tendency is the secondary hyperparathyroidism, which can cause pathologic fractures, bone deformities and reduction of life expectancy for patients. In conclusion, the secondary hyperparathyroidism, must be further studied in future investigations on clinical and laboratory studies to clarify the pathophysiology of this complication, besides the use of therapeutic proceedings and prophylaxis that increase life expectancy and quality in patients with CKD.
Keywords
Chronic kidney diseases; Hyperparathyroidism secondary; Risk factors
REFERÊNCIAS
- National Kidney Foundation. K/DOQI. Diretrizes de Prática Clínica para Doença Renal Crônica: Resumo Executivo. New York, 2002.
- Riella MC. Princípios de Nefrologia e Distúrbios Hidroeletrolíticos. 4a edição. Rio de Janeiro, Editora Guanabara Koogan 2003.
- Levey AS, Atkins R, Coresh J, Cohen EP, Collins AJ, Eckardt KU, et al. Chronic kidney disease as a global public health problem: approaches and initiatives – a position statement from Kidney Disease Improving Global Outcomes. Kidney Int. 2007 Aug;72(3): 247-59.
- Sesso R, Gordan P. Dados disponíveis sobre a doença renal crônica no Brasil. J Bras Nefrol Vol XXIX – nº 1, Março de 2007.
- Barros E, Manfro RC, Thomé FS, Gonçalves LF. Nefrologia: Rotinas, Diagnóstico e Tratamento. 3º edição, Porto Alegre, Artmed 2006.
- Vassalotti JA, Stevens LA, Levey AS. Testing for chronic kidney disease: a position statement from the National Kidney Foundation. Am J Kidney Dis. 2007 Aug;50(2):169-80.
- Moreira B, Fernandes PFCBC, Monte FS, Galvão RIM, Martins AMC. Conhecimento sobre o tratamento farmacológico em pacientes com doença renal crônica. Rev Bras Cienc Farm 2008 Abr-Jun;44(2):315-25.
- Soares AA. Desempenho da taxa de filtração glomerular estimada pelas fórmulas do estudo MDRD e equação quadrática mayo em adultos saudáveis. Universidade Federal do Rio Grande do Sul. Faculdade de Medicina. Programa de Pós-Graduação em Ciências Médicas: Endocrinologia. Acessível em http://www.lume.ufrgs.br/handle/10183/15926.
- Romão Jr JE. Doença renal crônica: definição, epidemiologia e classificação. J Bras Nefrol. 2004;26 Suppl 1:1-3.
- Hauser AB, Stinghen AE, Kato S, Bucharles S, Aita C, Yuzawa Y, et al. Characteristics and causes of immune dysfunction related to uremia and dialysis. Perit Dial Int. 2008 Jun;28 Suppl 3:S183-7.
- Porto CC. Vademecum de Clinica Médica. 2ª ed. Goiânia: Guanabara Koogan, 2008/2009, v. 01.
- Draibe SA, Ajzen H. Insuficiência renal crônica: problemas e soluções, NIEPEN e Fundação IMEPEN, Juiz de Fora, 2004.
- Cardoso FNC, Yanaguizawa M, Taberner GS, KubotaI ES, Fernandes ARC, Natour J. Contribuição da avaliação radiológica no hiperparatireoidismo secundário. Rev. Bras. Reumatol. 2007 Jun;47(3): 207-11.
- Lehmkuhl A, Maia AJM, Machado MO. Estudo da prevalência de óbitos de pacientes com doença renal crônica associada à doença mineral óssea. J Bras Nefrol. 2009;31(1):10-7.
- Prospero JD, Baptista PP, Amary MF, Santos PP. Paratireoides: estrutura, funções e patologia. Acta ortop. bras., 2009;17(2):53-7.
- Canto M, Lauand GCT. Deficiência de vitamina D e fatores determinantes dos níveis plasmáticos de 25-hidroxivitamina D. Brasília méd. 2008;45(3):208-17.
- Leal STC. Correlação dos achados histopatológicos das glândulas paratireoides com as características clínicas e laboratoriais de pacientes com hiperparatireoidismo secundário à insuficiência renal crônica submetidos à paratireoidectomia. Dissertação de Mestrado. Programa de Pós-graduação em Endocrinologia, Departamento de Clinica Médica, Universidade Federal do Rio de Janeiro, 2007.
- Gracitelli ECM, Vidoris AAC, Luba R, Castro, LM. Paratormônio e osteoporose: bases fisiológicas para utilização do PTH no tratamento da osteoporose. Arq Bras Endocrinol Metab. 2002 June; 46(3):215-20.
- Gonçalves CDM, Rodrigues SSA. Cirurgia do Hiperparatireoidismo. Rev. Col. Bras. Cir. 2002 June;29(3):166-76.
- Houssay BA, Cingolani EH. Fisiologia Humana de Houssay. 7ª edição. Porto Alegre. Editora Artmed 2004.
- Cobayashi F, Lopes AL, Taddei JAAC. Densidade mineral óssea de adolescentes com sobrepeso e obesidade. Jornal de Pediatria. 2005;81(04):337-42.
- Guyton AC, Hall JE. Tratado de Fisiologia Médica. 11ª edição. Rio de Janeiro. Editora Elsevier 2006.
- Grudtner VS, Weingrill P, Fernandes AL. Aspectos da absorção no metabolismo do cálcio e vitamina D. Rev Bras Reumatol. 1997;37:143-51.
- Lee JH, O’Keefe JH, Bell D, Hensrud DD, Holick MF. Vitamin D deficiency an important, common, and easily treatable cardiovascular risk factor. J Am Coll Cardiol. 2008 Dec 9;52(24): 1949-56.
- Sampaio EA, Lugon JR, Barreto CF. Fisiopatologia do hiperparatireoidismo secundário. J Bras Nefrol. 2008;30(Supl 1):6-10.
- Gomes EMS, Nunes RC, Lacativa PGS, Almeida MHF, Franco FM, Leal CTS, et al. Ectopic and extranumerary parathyroid glands location in patients with hyperparathyroidism secondary to end stage renal disease. Acta Cir Bras. 2007 Mar-Apr;22(2):105-9.
- Levin A, Bakris GL, Molitch M, Smulders M, Tian J, Williams LA, et al. Prevalence of abnormal serum vitamin D, PTH, calcium, and phosphorus in patients with chronic kidney disease: results of the study to evaluate early kidney disease. Kidney Int. 2007 Jan;71 (1):31-8.
- Peters BSE, Jorgetti V, Martini LA. Influência do hiperparatireoidismo secundário grave no estado nutricional de pacientes com insuficiência renal crônica. Rev. Nutr. 2006;19(1):111-8.
- Brazilian Guidelines for Bone and Mineral Disorders in CKD. Diretrizes Brasileiras de Prática Clínica para o Distúrbio Mineral e Ósseo na Doença Renal Crônica. J Bras Nefrol 2008;30(Supl 2):2-3. Disponível em http://dx.doi.org/10.1590/S0101-28002011000200021
- Acceta P, Cruz SAE, Lugon RJ. Paratireoidectomia subtotal no tratamento do hiperparatireoidismo secundário à doença renal crônica. J Bras Nefrol Volume XXVIII – nº 2 – Junho de 2006.
- Lorenzo V, Portillo MR, García RP, Andía JBC. De la osteodistrofia renal a las alteraciones del metabolismo óseo y mineral asociado a la enfermedad renal crónica: Evolución de un concepto. Nefrologia. 2007; 27(5):527-33.
- Goodman MG, William G. Atualização sobre os ensaios clínicos com cinacalcete no hiperparatireoidismo secundário associado à doença renal crônica. J Bras Nefrol 2008;30(Supl 1):44-50.
- Martin KJ, González EA. Metabolic bone disease in chronic kidney disease. J Am Soc Nephrol. 2007 Mar;18(3):875-85.
- Cannata-Andía JB, Fernández-Martín JL, Zoccali C, London GM, Locatelli F, Ketteler M, et al. Current management of secondary hyperparathyroidism: a multicenter observational study (COSMOS). J Nephrol. 2008 May-Jun;21(3):290-8.
- Bover J, Canal C, Marco H, Fernandez-Llama P, Bosch R, Ballarín J. Diagnostic procedures and rationale for specific therapies in chronic kidney disease-mineral and bone disorder. Contrib Nephrol. 2008; 161:222-33.
- Moe S, Drüeke T, Cunningham J, Goodman W, Martin K, Olgaard K, et al; Kidney Disease: Improving Global Outcomes (KDIGO). Definition, evaluation, and classification of renal osteodystrophy: a position statement from Kidney Disease: Improving Global Outcomes (KDIGO). Kidney Int. 2006 Jun;69(11):1945-53.
- CL Neves, MR Custódio, KR Neves, RMA Moysés, V Jorgetti. O hiperparatireoidismo secundário e a doença cardiovascular na doença renal crônica. J Bras Nefrol. 2008;30 (Supl 1):18-22.
- Rahimian M, Sami R, Behzad F. Evaluation of secondary hyperparathyroidism in patients undergoing hemodialysis. Saudi J Kidney Dis Transpl. 2008 Jan;19(1):116-9
- Cibulka R, Racek J. Metabolic disorders in patients with chronic kidney failure. Physiol Res. 2007;56(6):697-705.
- Isaia GC, Tamone C, Ravazzoli M. Fractures and chronic renal insufficiency. G Ital Nefrol. 2008 Jan-Feb;25(1):57-65. [Article in Italian]
- Wesseling K, Bakkaloglu S, Salusky I. Chronic kidney disease mineral and bone disorder in children. Pediatr Nephrol. 2008 Feb;23(2):195-207
- Buarqub MA, Nabulsi MF, Shafeh TA. Prevalence and pattern of renal osteodystrophy in chronic hemodialysis patients: a cross sectional study of 103 patients. Saudi J Kidney Saudi J Kidney Dis Transpl. 2006 Sep;17(3):401-7.
- Raggi P, Kleerekoper M. Contribution of bone and mineral abnormalities to cardiovascular disease in patients with chronic kidney disease. Clin J Am Soc Nephrol. 2008 May;3(3):836-43.
- Lacativa PG, Franco FM, Pimentel JR, Patrício Filho PJ, Gonçalves MD, Farias ML. Prevalence of radiological findings among cases of severe secondary hyperparathyroidism. Sao Paulo Med J. 2009 May; 127(2):71-7.
- Kulak CA, Borba VZ, Kulak Júnior J, Shane E.Transplantation osteoporosis. Arq Bras Endocrinol Metabol. 2006 Aug;50(4):783-92.
- Komaba H, Tanaka M, Fukagawa M. Treatment of chronic kidney disease – mineral and bone disorder (CKD-MBD). Intern Med. 2008;47(11):989-94.
- Smith DH, Johnson ES, Thorp ML, Yang X. Adherence to K/doqi bone metabolism guidelines. J Ren Nutr. 2009 Jul;19(4):334-42.
- Al Baddr W, Martin KJ. Role of bone biopsy in renal osteodystrophy. Saudi J Kidney Dis Transpl. 2009 Jan;20(1):12-9.
- Leonard MB. A structural approach to skeletal fragility in chronic kidney disease. Semin Nephrol. 2009 Mar;29 (2): 133-43.
- Ott SM. Bone histomorphometry in renal osteodystrophy. Semin Nephrol. 2009 Mar;29(2):122-32.
- Ferreira A, Frazão JM, Monier-Faugere MC, Gil C, Galvao J, Oliveira C, et al; Sevelamer Study Group. Effects of sevelamer hydrochloride and calcium carbonate on renal osteodystrophy in hemodialysis patients. J Am Soc Nephrol. 2008 Feb;19(2):405-12.
- Nickolas TL, Leonard MB, Shane E. Chronic kidney disease and bone fracture: a growing concern. Kidney Int. 2008 Sep;74(6):721-31.
- Saliba W, El-Haddad B. Secondary hyperparathyroidism: pathophysiology and treatment. J Am Board Fam Med. 2009 Sep-Oct;22(5):574-81.
- Martins MTS, Silva LF, Travessa IEM. Prescrição de Quelantes de Fósforo e Calcitriol para Pacientes em Hemodiálise Crônica. Rev. Assoc. Med. Bras. vol.55 nº 1. São Paulo 2009.
- Cruz J, Barros TR, Kirsztajn MG, Cruz MMH. Atualidades em Nefrologia. Editora Sarvier, 2008. São Paulo.
- Brandi L. 1alpha(OH)D3 One-alpha-hydroxy-cholecalciferol–an active vitamin D analog. Clinical studies on prophylaxis and treatment of secondary hyperparathyroidism in uremic patients on chronic dialysis. Dan Med Bull. 2008 Nov;55(4):186-210.
Correspondência
Aline Borsato Hauser
Av. Pref. Lothario Meissner, 632 – Campus Jardim Botânico
Setor de Ciências da Saúde – Prédio Azul da Farmácia
Laboratório Escola de Análises Clínicas
80210-170 – Curitiba, PR