Ocorrência de linfoma de Burkitt secundário a mieloma múltiplo em paciente tratado com protocolo CyBorD: um relato de caso
Occurrence of Burkitt lymphoma secondary to multiple myeloma in a patient treated with CyBorD protocol: a case report
Guilherme Dienstmann1
Carine Carla da Rosa2
Ana Paula Franz2
Matheus Leite Ramos de Souza3
Denise Ramos de Almeida2
Fernanda Marcante Carlotto2
Válber de Freitas Matias4
Rodrigo Ribeiro e Silva3
Samuel Ricardo Comar5
1Universidade Sociedade Educacional de Santa Catarina – UniSociesc. Blumenau-SC, Brasil.
2Hospital de Clínicas de Passo Fundo – HCPF. Passo Fundo – RS, Brasil.
3Universidade da região de Joinville – Univille. Joinville-SC, Brasil.
4Hemocentro de Alagoas – Hemoal. Maceió-AL, Brasil.
5Complexo Hospital de Clínicas da Universidade Federal do Paraná – UFPR. Curitiba – PR, Brasil.
Instituição: Universidade Sociedade Educacional de Santa Catarina – UniSociesc. Blumenau-SC, Brasil.
Recebido em 05/11/2020
Aprovado em 14/01/2021
DOI: 10.21877/2448-3877.20210001
INTRODUÇÃO
O mieloma múltiplo (MM) é uma neoplasia maligna caracterizada pela infiltração de plasmócitos clonais (células B) na medula óssea e, mais raramente, em sítios extramedulares, causando danos a vários órgãos, o qual é tipicamente resumido pela sigla CRAB (hipercalcemia, falência renal, anemia e lesões osteolíticas da medula óssea). O MM é responsável por aproximadamente 10% de todas as neoplasias hematológicas e, além de ser uma doença incurável, é a causa de cerca de 20% das mortes por neoplasias hematológicas e por 2% das mortes por todos os tipos de câncer. Existem evidências de que sua evolução se dá a partir de dois eventos: primeiramente um estágio pré-maligno assintomático, de proliferação de células plasmáticas clonais, denominado gamopatia monoclonal de significado indeterminado (MGUS) e um segundo evento que ocorre aleatoriamente e não por meio de dano cumulativo e que resulta no acúmulo de células tumorais que progridem para MM. Vários eventos de progressão podem atuar como esse segundo evento, incluindo tanto alterações genéticas quanto alterações no microambiente da medula óssea. Aproximadamente, 60% dos pacientes que possuem cariótipos anormais apresentam translocações cromossômicas em 14q32, como t (11; 14) (q13; q32) ou t (8; 14) (q24; q32).(1-3)
Já o linfoma de Burkitt (LB) é um linfoma não Hodgkin de células B indiferenciadas, altamente agressivo, com maior prevalência no gênero masculino, no qual praticamente todos os casos estão associados a uma translocação entre o braço longo do cromossomo 8, local do gene c-MYC (cellular-Myelocytomatosis) 8q24 e um dos três genes das imunoglobulinas (Ig): (i) gene da cadeia pesada de Ig no cromossomo 14 – resultando em t (8; 14) (q24; q32) encontrado em 80% dos casos de LB; (ii) gene da cadeia leve kappa no cromossomo 2 – resultando em t (2; 8) (p11; q24) encontrado em 15% dos casos LB; e (iii) gene da cadeia leve lambda no cromossomo 22 – resultando em t (8; 22) (q24; q11) encontrado em 5% dos casos de LB. Três formas clínicas distintas de LB são reconhecidas: endêmico, esporádico e associado à imunodeficiência (pacientes HIV/SIDA, ou que se encontram imunocomprometidos). Embora sejam histologicamente idênticos e com comportamento clínico semelhante, existem diferenças na epidemiologia, na apresentação clínica e nas características genéticas entre as três formas.(4,5)
O mieloma múltiplo e os linfomas não Hodgkin são doenças linfoproliferativas e a ocorrência de ambas as doenças em um mesmo paciente, seja concomitante ou secundária, é muito rara e apenas poucos casos foram descritos previamente.(6,7) Neste relato de caso, nós reportamos o caso de um paciente inicialmente diagnosticado com MM e que foi subsequentemente diagnosticado com LB durante a progressão do MM e que estava sendo tratado conforme protocolo CyBorD. Simultaneamente ao diagnóstico de LB, o paciente desenvolveu síndrome de lise tumoral e foi a óbito. Devido ao fato raro de um mesmo paciente apresentar MM e LB em tão curto período de tempo, este relato foi restrito a somente um único paciente, contudo acreditamos que este caso seja um exemplo representativo da transformação de um clone de células de MM para um linfoma de Burkitt.
RELATO DO CASO
Paciente masculino, 37 anos, negro, senegalês, HIV positivo com carga viral indetectável, em tratamento há três anos. No dia 28/01/2020, procurou emergência devido a febre, náuseas, vômitos, parestesias em boca e mandíbula e dor nos ossos. Avaliação inicial revelou lesão renal aguda não oligúrica; hipercalcemia, anemia normocítica/normocrômica e radiografia de crânio com característica em sal e pimenta. Os exames laboratoriais apresentaram os seguintes resultados: ureia 144 mg/dL (VR 15 a 40 mg/dL), creatinina 4,57 mg/dL (VR 0,70 a 1,30 mg/dL), cálcio total 15,24 mg/dL (VR 8,9 a 10,1 mg/dL), cálcio iônico 1,85 mg/dL (VR até 1,00 a 1,20 mmol/L), hemoglobina 9,9 g/dL (VR 12,8 a 17,8 g/dL), LDH 2.009 U/L (VR 122 a 222 U/L), b2-microglobulina 14,09 mg/L (VR 1,00 a 2,40 mg/L), albumina 2,9 g/dL (3,5 a 5,0 g/dL), ausência de pico de proteína monoclonal na eletroforese de proteínas e na imunofixação, IgG 1457 mg/dL (VR 650 a 1600 mg/dL), IgM 114 mg/dL (VR 50 a 300 mg/dL), IgA 340 mg/dL (VR 40 a 350 mg/dL) e cadeia leve kappa 311 mg/dL (VR 170 a 370 mg/dL). A amostra de urina de 24 horas apresentou cadeia leve kappa 11,700 mg/dL (VR até 0,710 mg/dL), cadeia leve lambda 2,970 mg/dL (VR até 0,390 mg/dL) e relação kappa/lambda 3,94 (VR 0,75 a 4,5). Nessa mesma amostra, a imunofixação demonstrou presença de proteína monoclonal kappa isolada. O mielograma evidenciou uma medula óssea hipocelular, com relação M:E aumentada 11,8:1 (VR 1,10:1 – 4,10:1) e com presença de 32% de plasmócitos. A imunofenotipagem resultou em granulócitos (67,0%), eosinófilos (3,2%), monócitos (2,7%), linfócitos (11,6%), eritroblastos (6,8%), plasmócitos (8,0%) e região de CD45-fraco (0,8%). A relação kappa/lambda manteve-se na população de linfócitos B, com presença de população clonal (8,0% dos eventos) expressando positividade para: CD81, CD138, CD38 fraco, CD117, CD56 heterogêneo, CD45 forte, com restrição de cadeia kappa e negatividade para CD19, CD20, CD27, CD28, CD56, CD103, CD10, CD13, CD5, CD200, CD22 e cadeia lambda, evidenciando assim, que a medula óssea encontrava-se infiltrada por plasmócitos displásicos (Figura 1), compatível com mieloma múltiplo, segundo classificação da OMS, 2017 e caracterizado como Estádio III, segundo o International Staging System (ISS). Foi iniciado, então, protocolo de quimioterapia CyBorD e Denosumabe para os ossos.
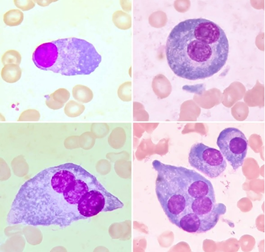
Figura 1. Plasmócitos displásicos (apresentando 2, 3 e 4 núcleos).
No dia 12/04/2020 (D1), após segundo ciclo de quimioterapia, em uso de sulfametoxazol + trimetoprima (1cp/dia) e aciclovir (3x/dia), paciente veio à emergência referindo epigastralgia de forte intensidade em aperto, associada a náuseas e vômitos (um episódio de hematêmese) e quadro de febre associada a dispneia aos moderados esforços. Paciente apresentou-se: desidratado; mucosas hipocoradas; PA 120/70; pulso 115 batimentos/minuto; saturando 96% em ar ambiente; temperatura de 37,8°C; abdome depressível, indolor, sem peritonismo e visceromegalias. Instalado acesso periférico em antebraço direito recebendo soroterapia a 40 mL/h e iniciado, então, tratamento para pneumonia adquirida na comunidade.
No dia 14/04/2020 (D3), após reavaliação, paciente relata desconforto respiratório e abdominal associado após episódios de diarreia e vômitos durante a noite. Exames laboratoriais evidenciaram: LDH 13.464 U/L (VR 122 a 222 U/L); ácido úrico 24,9 mg/dL (VR 2,7 a 6,1 mg/dL); lactato 101,30 mg/dL (VR 3,60 a 18,02 mg/dL); creatinina 2,75 mg/dL (VR 0,55 a 1,02 mg/dL); ureia 45 mg/dL (VR 15 a 40 mg/dL); potássio 4,7 mmol/L (VR 3,6 a 5,2 mmol/L); TGO 431 U/L (VR 8 a 48 U/L); TGP 59 U/L (VR 7 a 55 U/L); proteína C reativa 209,21 mg/L (VR até 3,00 mg/L); VHS 72 mm (VR até 20 mm); leucócitos 29.660 mm3 (VR 3.600 a 11.000/µL), com observação de presença de 58% de linfócitos anormais de tamanho médio, moderada relação núcleo/citoplasma, apresentando intensa basofilia e vacuolização nuclear e citoplasmática (Figura 2); hemoglobina 8,6 mg/dL (VR 12,8 a 17,8 mg/dL), com observações de policromasia (++), eritroblastos (8%) e presença de rouleaux eritrocitário; e plaquetas 27.000/µL (VR 140.000 a 360.000/µL).
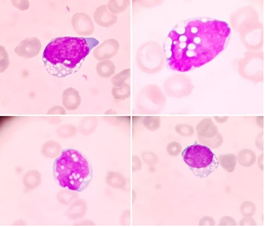
Figura 2 – Linfócitos anormais apresentando vacuolização nuclear e citoplasmática
No dia 15/04/2020 (D4), devido a quadro de instabilidade hemodinâmica, paciente foi levado à unidade de terapia intensiva. Realizado acesso venoso central duplo-lúmen em femoral esquerda, recebendo noradrenalina 3 mL/h, e acesso venoso periférico em membro superior esquerdo, recebendo midazolam (10 mL/h) + fentanil (3 mL/h) + bicarbonato (60 mL/h). Paciente hipoglicêmico, administrado 4 ampolas de glicose, bicarbonato 250 mL. Apresentou agitação psicomotora, administrado uma dose de ataque de midazolam 15 mg + brometo de pancurônio. Sob a hipótese de lise tumoral e progressão tumoral, foi realizada nova Imunofenotipagem, a qual apresentou: população clonal linfoide B (70%); antígeno de núcleo: TdT negativo; antígenos de citoplasma: CD79a+, Bcl-2 e CD3 negativos; antígenos de membrana: CD19+/++, CD20+/++, CD45+/++, CD38+/++, CD10+, CD79b-/++ (78%) positivos; CD34, CD7, CD5, CD3, CD23, CD200, CD43, CD11c, cadeia lambda, negativos. Concluindo assim que a análise imunofenotípica apresentava-se compatível com linfoma linfoblástico B e o imunofenótipo sugeria o subtipo Burkitt. A análise citogenética não foi realizada.
Na madrugada do dia 16/04/2020 (D5), o paciente evoluiu com insuficiência respiratória, parada cardiorrespiratória em assistolia e óbito.
DISCUSSÃO
Embora exista um risco aumentado de evolução para leucemia mieloide aguda e síndromes mielodisplásicas após tratamento para MM,(7) a transformação em LB é incomum, com raros casos relatados e ainda muito a se compreender sobre a relação entre o MM e subsequente evolução para leucemia linfoblástica aguda (LLA).(8,9)
Neste relato de caso, nosso paciente preencheu inicialmente todos os critérios para MM, com presença de cadeias leves kappa e lambda aumentadas na urina, além de plasmocitose medular com plasmócitos displásicos, de tamanho irregular, alguns agrupados e outros com inclusões de imunoglobulinas. Pouco mais de dois meses após o diagnóstico de MM, o paciente evoluiu para uma transformação medular que resultou em um linfoma linfoblástico B compatível com LB e para um quadro de síndrome de lise tumoral que culminou em seu óbito. Pacientes com MM podem se apresentar com características clínicas variáveis e em diferentes estágios da doença. MM é uma doença na qual a grande maioria dos pacientes apresenta uma proteína monoclonal no soro e/ou urina, que leva ao conceito predominante de que o MM seja monoclonal ao nível celular. Embora os linfomas sejam, geralmente, neoplasias dos tecidos linfáticos, um número substancial de linfomas surge em outros tecidos, como a medula óssea, na qual a incidência dos linfomas não Hodgkin é de, aproximadamente, 65% na ocasião do diagnóstico.(8)
A ocorrência de ambos, MM e LB, em nosso paciente, implica na possibilidade de influência de vários mecanismos fisiopatológicos, dentre os quais a evolução de uma doença neoplásica para outra, no caso, o MM evoluir para um LB. A comparação dos imunofenótipos de ambas as populações clonais estudadas em nosso paciente poderia fornecer indícios para alcançarmos uma conclusão significativa, contudo, tais imunofenótipos não foram intercambiáveis e estes achados sugerem que não houve transformação do clone de MM para o clone de LB. Outros casos anteriormente relatados e nos quais foram empregadas técnicas moleculares para comparação de clones, também não mostraram relação clonal da LLA com o MM inicial.(9,10)
Estudos destacaram a desregulação de c-MYC como um possível evento molecular importante na oncogênese, tanto do MM quanto do LB, a qual leva a um aumento de sua expressão e, consequentemente, à proliferação neoplásica. O LB está classicamente associado à translocação (8; 14) (q24; q32), contudo ela também foi observada no pequeno linfoma de células não clivadas, no linfoma imunoblástico e, menos frequentemente, no MM, no qual os cariótipos anormais são observados em cerca de 30%-50% dos casos associados a cursos agressivos de doenças.(4) Estudos realizados em plasmocitomas de camundongos e linhagens celulares de mieloma humano destacaram a desregulação de c-MYC como um possível evento molecular importante na oncogênese do MM. Entretanto, esses rearranjos podem aparecer tardiamente na história natural do MM humano, representando eventos oncogenéticos secundários e não primários. Os rearranjos de c-MYC foram observados significativamente mais frequentemente em pacientes com níveis de b2-microglobulina acima de 3 mg/L,(3,11) como em nosso paciente (14,09 mg/L).
Já foram relatados vários casos de MM com a variante tipo Burkitt t (8; 22) (q24; q11)(3,5,12-18) (e considerando que, aproximadamente, 60% dos pacientes com MM que possuem cariótipos anormais apresentam translocações cromossômicas em 14q32, como t (11; 14) (q13; q32) ou t (8; 14) (q24; q32)(1-3) e que translocações variantes, como, por exemplo, t(2; 8)(p12; q24) ou t(8; 22)(q24; q11), também são observadas especificamente no linfoma de Burkitt,(1-4) levantamos a hipótese de que o nosso paciente apresentava a translocação variante do tipo Burkitt, t (8; 22) (q24; q11), promovendo assim ativação e desregulação do gene c-MYC, o que implicou na progressão do MM. Contudo, para corroborar tal hipótese, exames citogenéticos confirmatórios seriam necessários, os quais não puderem ser realizados em virtude do óbito do paciente.
A transformação maligna de MM, como a apresentada neste relato, foi raramente descrita, contudo, durante as últimas décadas, tem-se observado maiores taxas de sobrevivência em pacientes com MM devido aos avanços nos tratamentos e nos cuidados com os pacientes. Essa taxa de sobrevivência aumentada tem permitido um aumento nas chances de serem observadas transformações do MM para linfomas malignos de alto grau. Devemos também considerar a hipótese de nosso paciente ter evoluído de MM para LB em decorrência do tratamento citotóxico, conforme já descrito por Wahlin, Roos & Horsten (1988),(19) ou necrose tumoral espontânea, que o fez evoluir para o quadro de síndrome de lise tumoral e óbito. A síndrome de lise tumoral observada em nosso paciente também pode ter ocorrido em virtude do tratamento com ciclofosfamida, bortezomib e dexametasona (CyBorD), conforme já descrito por Reeder et al (2009).(20)
Outro mecanismo possível seria que ambas as neoplasias sejam diferentes manifestações do clone neoplásico original e que o aparecimento das duas neoplasias em tão curto intervalo de tempo representaria diferentes manifestações de um único clone que afetou os precursores das células B.(8,9). Embora rara, existe ainda a possibilidade de ocorrer as duas doenças ao mesmo tempo.(21) Este caso oferece apoio adicional para a teoria de transformação maligna de MM para LB. Oportunidades para estudar e compreender a história natural única destas transformações são raras. Nesse sentido, recomendamos futuras comparações multicêntricas de casos similares num esforço amplo para uma melhor definição desse processo patológico.
Abstract
Introduction: After treatment for Multiple Myeloma (MM), transformation into burkitt’s lymphoma (LB) is uncommon, with rare cases reported.
Objective: to report a rare case of LB secondary to MM, in a patient treated with the CyBorD protocol. Case report: We describe a case of a 37-year-old man, black, Senegalese and HIV positive, who in just over 2 months after the diagnosis of MM evolved to a medullary transformation that resulted in a B lymphoblastic lymphoma compatible with LB. Conclusion: This case report demonstrates the possibility of malignant transformation from MM to BL, thus encouraging future multicentric comparisons of similar cases in a broad effort to better define this pathological process.
Keywords
Multiple myelome; Burkitt lymphoma; CyBorD protocol
REFERÊNCIAS
- Kim SH, Kim JH, Lee DM, Lee S, Oh SY, Kwon HC, et al. Comparison between conventional cytogenetics and interphase fluorescence in situ hybridization (FISH) for patients with multiple myeloma. Korean J Hematol, 2009;44(1):14-21. https://doi.org/10.5045/kjh.2009.44.1.14
- Swerdlow SH, Campo E, Harris NL, et al, eds. World Health Organization classification of tumours of haematopoietic and lymphoid tissues. 4th ed. Lyon, France: IARC Press, 2008.
- Taniwaki M, Nishida K, Takashima T, Nakagawa H, Fujii H, Tamaki T, et al. Nonrandom chromosomal rearrangements of 14q32.3 and 19p13.3 and preferential deletion of 1p in 21 patients with multiple myeloma and plasma cell leukemia. Blood 1994;84(7):2283-2290.
- Lee KH, Lee YK, Park JY, et al. A case report of t(8;22) with additional chromosomal defects in bone marrow involvement of B cell type diffuse large cell lymphoma. Korean J Clin Pathol 1998;18:525-528.
- Yamamoto K, Hamaguchi H, Nagata K, Taniwaki M. A variant Burkitt-type translocation (8;22)(q24;q11) in multiple myeloma. Report of a new case and review of the literature. Cancer Genet Cytogenet 1998;104:98-103. doi: 10.1016/s0165-4608(97)00458-5.
- Brouet JC, Fernand JP, Laurent G, Grance MJ, Chevalier A, Jacquillat C, Seligmann M. The association of chronic lymphocytic leucemia and multiple myeloma: A study of eleven patients. Br J Haematol. 1985 Jan;59(1):55-66.. doi: 10.1111/j.1365-2141.1985.tb02963.x
- Mailankody S, Pfeiffer RM, Kristinsson SY, Korde N, Bjorkholm M, Goldin LR, et al. Risk of acute myeloid leucemia and myelodysplastic syndromes after multiple myeloma and its precursor disease (MGUS). Blood, 2011;118:4086-4092. doi: 10.1182/blood-2011-05-355743.
- Lee GC, Hong JS, Lee KH, Kim SB, Kim SW, Suh CW, et al. A case of coincidente multiple myeloma and non-Hodgkin’s lymphoma. Korean J Inten Med, 1994;9(2): 113-115. doi: 10.3904/kjim. 1994.9.2.113
- Grau E, Soler J, Puig JÁ, Brunet S, Pujol-Moix N, Rodriguez JL, et al. Coincident multiple myeloma and non-Hodgkin’s lymphoma with 2 serum monoclonal imunoglobulins. Acta Haematol, 1986;75:183-185. doi: 10.1159/000206117
- Kyle RA, Rajkumar SV. Criteria for diagnosis, staging, risk stratification and response assessment of multiple myeloma. Leukemia, 2009;23:3-9.
- Avet-Loiseau H, Gerson F, Magrangeas F, et al. Rearrangements of the c-myc oncogene are present in 15% of primary human multiple myeloma tumors. Blood 2001;98:3082-3086.
- Solé F, Woessner S, Acín P, et al. Burkitt’s type translocation in multiple myeloma. Leuk Res 1994;18:671-3.
- Sawyer JR, Waldron JA, Jagannath S, Barlogie B. Cytogenetic findings in 200 patients with multiple myeloma. Cancer Genet Cytogenet 1995;82:41-9.
- Look RM, Lim SW, Schreck RR, et al. Burkitt translocation (8;22)(q24;q11) in a patient with multiple myeloma. Cancer Genet Cytogenet 1995;82:100-2.
- Mugneret F, Sidaner I, Favre B, et al. Der(16)t(1;16)(q10;p10) in multiple myeloma: a new non-random abnormality that is frequently associated with Burkitt’s-type translocations. Leukemia 1995;9:277-81.
- Calasanz MJ, Cigudosa JC, Odero MD, et al. Cytogenetic analysis of 280 patients with multiple myeloma and related disorders: primary breakpoints and clinical correlations. Genes Chromosomes Cancer 1997;18:84-93.
- Sugiyama A, Nakabayashi H, Kondo M, Tominaga T, Shinohara K. Multiple myeloma with variant type translocation, t(8;22)(q24; q11.2). Rinsho Ketsueki 2009;50:29-33. leukemia. Blood 1994;84:2283-90.
- Kim H., Moon H-W, Hur M, Yun Y-M, Park C-M, Lee M. Variant Burkitt-type translocation (8;22)(q24;q11) in plasma cell myeloma. Korean J Hematol 2011;46:135-8.
- Wahlin A, Roos G, Hörnsten P. Multiple Myeloma-Transformation to High-Grade Malignant Lymphoma, Acta Oncologica, 27:4;411-413, 1988.
- Reeder CB, Reece DE, Kukreti V, et al. Cyclophosphamide, bortezomib and dexamethasone (CyBorD) induction for newly diagnosed multiple myeloma: High response rates in a phase II clinical trial. Leukemia, 2009;23(7):1337-1341.
- Khade P, Devarakonda S. Coexisting multiple myeloma, lymphoma, and non-small cell lung câncer: a case report and review of the literature. Int Med Case Rep J, 2017;10:373-376.
Correspondência
Guilherme Dienstmann
Rua João Alexandre de França, 349
89225-140 – Joinville-SC, Brasil