Aplicação clínica e laboratorial do teste da mistura na investigação de prolongamentos nos exames de hemostasia
Clinical and laboratory application of the mixing test in the investigation of prolonged hemostasis testing
Larissa Aparecida Vieira1, Cesar Augusto Leite2, Samuel Ricardo Comar3
1 Complexo Hospital de Clínicas da Universidade Federal do Paraná, Residência Integrada Multiprofissional em Atenção Hospitalar – Área de Oncologia e Hematologia. Curitiba, PR, Brasil.
2 Centro de Hematologia e Hemoterapia do Paraná (HEMEPAR), Serviço de produção de hemocomponentes. Curitiba, PR, Brasil.
3 Complexo Hospital de Clínicas da Universidade Federal do Paraná, Laboratório de Hematologia. Curitiba, PR, Brasil.
Recebido em 18/09/2022
Aprovado em 08/12/2022
DOI: 10.21877/2448-3877.202200072
INTRODUÇÃO
A hemostasia é um complexo e intrincado processo que impede, em tempo hábil, o surgimento demasiado de sangramentos e a formação excessiva de coágulos quando uma lesão vascular ocorre. Seu principal objetivo é selar a área lesionada enquanto ocorre a regeneração dos tecidos e vasos sanguíneos adjacentes. Para isso, requer a ação combinada de vasos, plaquetas e fatores plasmáticos da coagulação, os quais atuam por meio de três fases intimamente ligadas: (a) hemostasia primária (vasoconstrição breve e intensa dos vasos sanguíneos e formação de um tampão plaquetário via interação entre plaquetas, fator de von Willebrand (FvW) e fatores teciduais para inicialmente interromper o sangramento); (b) hemostasia secundária (coagulação do sangue por meio da formação de uma malha de fibrina polimerizada que age como uma cola para estabilizar e manter o coágulo unido); e (c) hemostasia terciária (fibrinólise do coágulo de fibrina à medida que a cicatrização e reparo se iniciam no ferimento).(1,2)
Os testes de triagem em hemostasia mais utilizados para a avaliação da hemostasia secundária são o tempo de protrombina (TP), o tempo de tromboplastina parcial ativada (TTPa) e o tempo de trombina (TT). Na perspectiva laboratorial, resultados de TP, TTPa e TT inesperadamente prolongados devem ser investigados por meio da utilização do teste da mistura (TM), o qual consiste, normalmente, na realização do respectivo teste de triagem após a mistura do plasma do paciente com um pool de plasmas normais (PPN) na proporção de 1:1.(3) Nesse sentido, este trabalho teve como objetivo atualizar o estado da arte a respeito do uso do TM em direcionar e elucidar as alterações hemostáticas, assim como seus benefícios para o laboratório e os clínicos.
MATERIAIS E MÉTODOS
A metodologia de pesquisa utilizada foi revisão bibliográfica narrativa, e as bases de dados utilizadas foram: PubMed, LILACS, BVS, SciELO, Science Direct e Portal da Capes. Também foram incluídos materiais disponíveis de forma física em bibliotecas e acervos. A busca foi realizada utilizando os seguintes termos “mixing test”, “activated partial thromboplastin time”, “prothrombin time”, “lupus anticoagulant”, “thrombin time”, “anti-Xa”, “heparin test”, “pool plasma normal”, “fator deficiency”, “acquired factor deficiency”, “clotting assay”, “coagulation cascade”, “hemostasis assays”, “fibrinogen”, “factor II”, “factor V”, “factor VII”, “factor VIII”, “factor IX”, “factor X”, “factor XI”, “factor XII”, “disseminated intravascular coagulation”, “Von Willebrand factor”. Foram incluídos artigos e documentos escritos em português e inglês, considerados relevantes sobre o tema.
RESULTADOS E DISCUSSÃO
Testes de triagem no laboratório de hemostasia
O TP é realizado utilizando tromboplastina cálcica completa, que mimetiza a ação do fator tecidual (FT), levando à ativação da coagulação por meio da via extrínseca, principal via que o TP avalia, além da via comum da coagulação. Cabe ressaltar que, em razão da variedade de preparados de tromboplastina disponíveis comercialmente, torna-se necessária a padronização dos resultados de TP a fim de possibilitar comparações interlaboratoriais. Nesse sentido, os fabricantes de tromboplastinas submetem seus produtos a uma comparação com tromboplastinas internacionais de referência, como as do Laboratório Internacional de Produtos Biológicos da OMS para obtenção do índice de sensibilidade internacional (ISI) da tromboplastina comercial. Com o ISI é possível calcular a relação normalizada internacional (RNI), que é o parâmetro que torna possível a comparação de resultados entre diferentes laboratórios, com diferentes tromboplastinas. O RNI é especialmente útil no controle da terapia com anticoagulantes orais antagonistas da vitamina K (AVK).(3-7)
Já para a realização do TTPa, a tromboplastina parcial (denominada parcial pois não contém fator tecidual como na tromboplastina completa do TP) é utilizada como fonte de fosfolipídios que substitui as plaquetas ativadas nas reações. Também são adicionados ativadores da coagulação como o ácido elágico, a sílica micronizada, a celite e o caulim, os quais auxiliam efetuando ativação máxima da coagulação, eliminando os efeitos variáveis do contato do sangue com as superfícies de vidro ou plástico dos tubos de coleta. O TTPa é utilizado principalmente como teste de triagem para a avaliação da via intrínseca da coagulação, mas também da via comum e os seus resultados são liberados em segundos e, assim como no TP, devem ser comparados ao valor do PPN.(4,8)
O TT mede o tempo de coagulação do plasma após a adição de trombina, avaliando, desta forma, a via final da cascata da coagulação, sendo útil como teste de triagem para deficiências de fibrinogênio e inibidores de trombina.(3-8)
Fundamentos dos testes da mistura
Na perspectiva laboratorial, resultados de TP, TTPa e TT inesperadamente prolongados devem ser investigados por meio da utilização do TM. O TM consiste, normalmente, na realização do teste de triagem após a mistura do plasma do paciente com o PPN na proporção de 1:1. O PPN contém todos os fatores de coagulação (FC) com atividade próxima a 100% do normal, resultando em uma mistura contendo um nível pelo menos ≥ 50% de tais FC, promovendo assim, teoricamente, a correção dos valores de TP e/ou TTPa nas deficiências de FC e a não correção na presença de diferentes tipos de inibidores de fatores de coagulação (IFC) no plasma do paciente (Figura 1).(9-12)
Os TM apresentam limitações e geralmente não são úteis como parte do acompanhamento de um efeito conhecido decorrente de terapia anticoagulante, exceto para diferenciar a terapia com antagonistas da vitamina K (correção no TM) da terapia com outros anticoagulantes (TM normalmente não corrige no caso de inibidores diretos da trombina ou heparina, a menos que a mistura traga o nível de heparina dentro da capacidade de neutralização de um reagente em particular).(3,11,13)
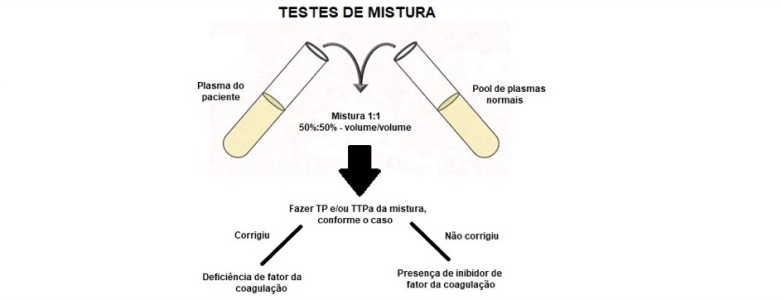
Figura 1
Execução dos testes de mistura
Notas: (i) os testes de triagem em hemostasia podem ser prolongados em razão da deficiência de fatores de coagulação ou presença de um inibidor da atividade normal de um fator de coagulação. (ii) os testes coagulométricos geralmente necessitam de uma atividade dos fatores de coagulação de 50% do normal para que o teste obtenha resultados normais. (iii) o grau de prolongamento de um TP ou TTPa depende do grau da deficiência de um determinado fator de coagulação, da força do inibidor de fator de coagulação e da sensibilidade dos reagentes à deficiência ou efeito inibidor.
Preparo do pool de plasmas normais
O preparo adequado do PPN é fundamental para a interpretação dos TM e deve seguir as recomendações da International Society on Thrombosis and Haemostasis (ISTH), do British Committee for Standards in Haematology (BCSH) ou do Clinical & Laboratory Standards Institute (CLSI). A ISTH, por exemplo, recomenda que sejam utilizadas pelo menos 40 amostras de indivíduos normais colhidas em tubo contendo citrato de sódio 3,2% e que a centrifugação seja realizada duas vezes para assegurar um residual mínimo de plaquetas inferior a 107 × mL-1 (10.000/µL) e um PPN com atividade dos FC próximas a 100%. Após o preparo in house do PPN, deve-se aliquotá-lo em pequenas quantidades (geralmente 1mL por tubo) e armazená-lo preferencialmente a -70°C por até 6 meses. É aceitável o armazenamento a -40°C por 3 meses e a -20°C por 2 semanas. Plasma normal liofilizado (comercial) também pode ser utilizado como PPN, contudo a vantagem de se preparar o próprio PPN consiste no fato de que este é mais representativo da população atendida pelo laboratório. Para a utilização nos TM, o PPN previamente preparado e congelado deve ser descongelado na temperatura de 37°C durante, no máximo, 5 minutos, devendo permanecer tampado para evitar evaporação. Intervalos de descongelamento maiores podem levar à inativação de FC e prejudicar a eficiência do PPN.(7,18-20)
Interpretação da “correção” e da “não correção” dos testes da mistura
A fim de confirmar se houve ou não correção do teste de triagem após a mistura com PPN, é fundamental uma correta e adequada interpretação dos resultados dos TM, a qual pode ser realizada de várias maneiras, sem que ainda haja consenso sobre qual delas é a melhor. Desta forma, cabe aos laboratórios escolherem e padronizarem a forma mais apropriada para a interpretação dos resultados dos TM.(3,8,11-15) Nenhuma abordagem de interpretação dos TM é 100% sensível e específica para distinguir deficiências ou inibidores de fatores de coagulação. É comum situações em que há sobreposição dos resultados dos TM de amostras com deficiência de fator versus inibidores, independentemente do método de interpretação utilizado.(3,11,13) As formas de interpretação da correção para os TM estão resumidas no Quadro 1.
Existem variações da técnica padrão de realizar os TM (mistura 1:1) como, por exemplo, utilizar mistura 4:1 com quatro partes de plasma do paciente para uma parte de PPN a fim de diminuir a interferência da diluição dos fatores de coagulação do paciente nos testes. A realização de uma etapa de incubação a 37oC durante 2 horas ajuda a verificar a presença de inibidores tempo e temperatura dependentes, como o caso dos inibidores do FVIII. Os TM estão sujeitos a variáveis pré-analíticas, analíticas e pós-analíticas. As pré-analíticas são punção; tipo de tubo de coleta; volume de amostra; ajuste da quantidade de citrato para hematócritos elevados (>55%); transporte; centrifugação; presença de medicamentos anticoagulantes; substâncias que se ligam a fosfolipídios in vitro e prolongam o TTPa, como anticorpos antilúpicos e antibióticos como oritavancina; presença de microcoágulos; hemólise; icterícia; lipemia; eficiência do PPN após seu preparo, a qual, idealmente, deve ser verificada em relação à normalidade dos tempos e atividades dos principais fatores de coagulação. As analíticas são sensibilidade dos reagentes aos fatores de coagulação; erros de diluição dos reagentes; tipo de analisador; erros de calibração; uso de diferentes lotes de PPN na mesma corrida. As pós-analíticas são realizar corretamente os cálculos e interpretações e reportar corretamente os resultados. Reconhecer e minimizar essas variáveis aumenta a confiabilidade dos resultados.(3,8,11-17)
Quadro 1
Métodos de interpretação do Teste da Mistura
| MÉTODOS | FORMA DE INTERPRETAÇÃO | |
| Utilizando o TC da mistura 1:1 (P/PPN) | Verificar se o TC da mistura 1:1 está acima do valor de referência estabelecido internamente para o TC do teste de triagem correspondente | Correção quando o TC da mistura está dentro dos valores de referência
Não correção quando o TC da mistura está acima dos valores de referência |
| Verificar se o TC da mistura 1:1 ultrapassa o valor estabelecido pela média dos valores obtidos de plasma normal ± 2 ou 3 DP | ||
| Utilizando o TC da mistura 1:1 (P/PPN) e o TC do PPN | Calcular relação entre o TC da mistura 1:1 e o TC do PPN do teste de triagem correspondente, utilizando como valor de corte, por exemplo, 1,1 ou 1,2 | Correção quando a relação está abaixo ou igual ao valor de corte preestabelecido
Não correção quando a relação está acima do valor de corte preestabelecido |
| Comparar a diferença entre TC da mistura 1:1 (P/PPN) e o TC do PPN do teste de triagem correspondente, considerando uma diferença preestabelecida, como por exemplo 5 segundos | Correção quando TC da mistura 1:1 varia dentro de 5 segundos em relação ao TC do PPN
Não correção quando TC da mistura 1:1 é >5 segundos em relação ao TC do PPN
|
|
| Comparar TC da mistura 1:1 (P/PPN) com os valores do 99° percentil da mistura 1:1 realizada em voluntários saudáveis (SAUDÁVEL/PPN) | Correção quando o TC da mistura está < 99° percentil da mistura 1:1 de voluntários saudáveis
Correção quando o TC da mistura está > 99° percentil da mistura 1:1 de voluntários saudáveis |
|
| Utilizando o TC da mistura 1:1, do PPN e do plasma do paciente | Calcular ICA (Índice de Rosner)14:
ICA = [(TC 1:1 mix – TC do PPN) / TC do paciente] × 100
onde ICA >11% ou >12% ou >15,6% são indicativos da presença de um inibidor, enquanto níveis <5% indicam deficiência |
Correção quando o ICA está abaixo ou igual ao valor de corte preestabelecido
Não correção quando o ICA está acima do valor de corte preestabelecido |
| Calcular porcentagem de correção (%C)15:
%C = [(TC do paciente – TC 1:1 mix) / (TC do paciente – TC do PPN)] × 100
onde %C de, por exemplo, >70% ou >75% indicam correção |
Correção quando a %C está abaixo ou igual ao valor de corte preestabelecido
Não correção quando a %C está acima do valor de corte preestabelecido |
|
| Verificar se TC da mistura 1:1 (P/PPN) corrige >50% da diferença entre o TC do paciente e o TC do PPN8 | Correção eficaz quando TC da mistura 1:1 é maior que 50% × (TC do paciente – TC do PPN)
Não correção quando TC da mistura 1:1 é menor que 50% × (TC do paciente – TC do PPN) |
|
Legenda: TC (tempo de coagulação em segundos para os testes de triagem de hemostasia, conforme o caso); P (plasma do paciente); PPN (pool de plasmas normais); ICA (índice de anticoagulante circulante); DP (desvio padrão).
APLICAÇÃO DOS TESTES DA MISTURA
Aplicação dos testes da mistura para verificar deficiência de fatores de coagulação
A deficiência de FC pode ser demonstrada pelos TM por meio da correção dos valores prolongados de TP e TTPa após mistura 1:1 com PPN. O prolongamento isolado do TP, seguido de correção, aponta a deficiência (quantitativa ou qualitativa) de FVII, mais frequente que deficiência de outros FC da via extrínseca. TP e TTPa prolongados, seguidos de correção, apontam para deficiência de FC da via comum ou de múltiplos FC, como ocorre em doenças hepáticas e coagulopatias de consumo. O prolongamento de TTPa, apenas, seguido de correção é sugestivo de hemofilia A ou B, não podendo descartar deficiência de outros FC da via intrínseca. Existem vários reagentes de TP e de TTPa disponíveis no mercado e cada um possui uma determinada sensibilidade frente às deficiências de FC. Isso significa que o nível abaixo do qual, por exemplo, o FVIII deve cair antes do TTPa prolongar não será o mesmo para todos os reagentes de TTPa e é provável que este nível não coincida com o limite inferior do valor de referência do laboratório. Contudo, de modo geral, os reagentes de TTPa devem ser sensíveis às deficiências de fatores VIII, IX e XI em concentrações de 35% a 40%.(9,21)
Aplicação dos testes da mistura para verificar presença de inibidores dos fatores de coagulação
Os IFC podem ser específicos ou inespecíficos. IFC específicos correspondem a autoanticorpos ou aloanticorpos do tipo IgG, IgM ou IgA dirigidos contra um determinado FC, o qual pode ser neutralizado ou ter sua velocidade de depuração acelerada devido ao complexo antígeno anticorpo formado. O exame confirmatório para se determinar a presença de IFC específicos é realizado por meio do método de Nijmegen, uma modificação do método de Bethesda. O teste consiste no uso de diluições seriadas de plasma deficiente de determinado FC, proveniente do paciente, misturado a um PPN tamponado (com concentração normal dos FC), a fim de quantificar a inibição da atividade do FC após incubação por tempo determinado. A diluição necessária para se atingir 25% a 75% da atividade do FC em questão, comparada ao controle, na mistura 1:1 de plasma do paciente e PPN tamponado corresponde ao título em unidades Bethesda. Quando múltiplos FC permanecem com atividade muito baixa, deve-se investigar a procedência das amostras e erros pré-analíticos, como coleta em ácido etilenodiamino tetra-acético (EDTA) ou amostra de soro, visto que a presença de múltiplos IFC é muito rara. Uma dosagem de potássio sérico pode ajudar a elucidar a presença de EDTA, enquanto uma dosagem de fibrinogênio pode identificar uma amostra de soro.(2,12,16,22)
ABORDAGEM LABORATORIAL
Quando ocorre prolongamento isolado do TP
O prolongamento do TP associado a um resultado de TTPa normal é característico de alterações que envolvem o FVII, entretanto também pode ocorrer na deficiência congênita de FX, FV, protrombina (FII) e deficiências de fibrinogênio (FI). O prolongamento isolado de TP também pode ser provocado pela presença de IFC específicos, anticorpos do tipo IgG contra FVII. Um teste da mistura de TP pode ser utilizado para dar sequência à investigação.(23)
Quando o teste da mistura do TP não corrige
Inibidor de FVII
A deficiência adquirida de FVII, resultante da presença de inibidores, ocorre de maneira pouco comum. Os inibidores específicos de FVII podem ser de duas naturezas e ambos se tratam de anticorpos IgG. Os anticorpos podem surgir em diferentes contextos, como sepse, doenças malignas, e relacionados ao uso de medicamentos como penicilinas, cefalosporinas, globulina antitimócitos e interleucinas. Também podem estar relacionados à presença de anticoagulante lúpico (AL). Foi reportada a presença de autoanticorpos dirigidos contra o FVII em alguns pacientes com histórico de síndrome do anticorpo antifosfolipídio (SAF). O diagnóstico da presença de inibidor específico contra FVII é realizado pela avaliação dos testes de triagem, sendo observado TP prolongado que não corrige após TM e através da realização do teste de Bethesda modificado para a sua quantificação (Figura 2).(22-26)
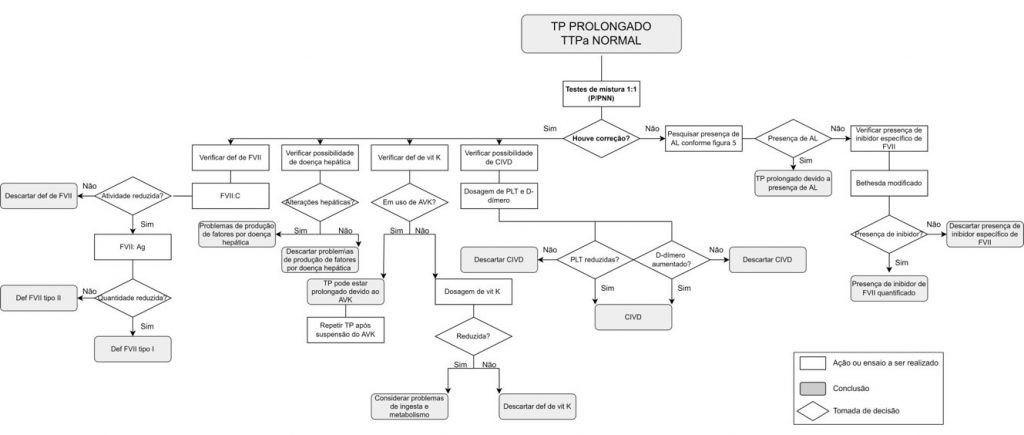
Figura 2
Prolongamento isolado de tempo de protrombina (TP)
Legenda: P/PPN (relação entre o tempo de coagulação do plasma do paciente e o pool de plasmas normais); def (deficiência); CIVD (coagulação intravascular disseminada); vit k (vitamina K); FVII:C (atividade de FVII); FVII:Ag (pesquisa de antígeno FVII, pesquisa quantitativa por ELISA ou imunoturbidimetria); AVK (antagonista da vitamina K); PLT (plaquetas); AL (anticoagulantes lúpicos).
Quando o teste da mistura do TP corrige
Deficiência hereditária de FVII
A deficiência congênita de FVII é uma alteração rara, com prevalência global de cerca de 1:500.000, e é caracterizada pela atividade do FVII:C inferior a 70%. Frequentemente é observada história familiar de sangramento quando FVII é < 30% e o diagnóstico, de modo geral, é feito ainda durante a infância. A deficiência pode ser do tipo I, na qual se observam alterações quantitativas, e do tipo II, na qual se observam alterações qualitativas. O diagnóstico consiste na avaliação do quadro clínico e laboratorial, observa-se TP prolongado, TM de TP com correção e atividade coagulante de FVII:C e antígeno FVII-Ag, alterados (Figura 2). É importante ressaltar que diferentes respostas podem ser observadas no TP devido à variedade dos preparados de tromboplastina e diferentes polimorfismos genéticos apresentados por pacientes com deficiência de FVII (FVII Padua, FVII Yamomoto). A quantificação de FVII:Ag pode ser realizada através de ensaios imunoturbidimétricos e imunoenzimáticos, contudo não deve ser utilizada como primeira escolha no diagnóstico, visto que não detecta alterações qualitativas funcionais, mas apenas quantitativas. Outro ponto a se considerar é que o FVII possui meia-vida de 4 a 6 horas, inferior aos outros fatores de coagulação, sendo o primeiro fator a diminuir em casos de deficiências combinadas, portanto é necessário repetição do exame caso a análise ultrapasse este tempo para confirmação de deficiência única ou combinada.(8,23,24,27)
Coagulação intravascular disseminada
A ocorrência de coagulação intravascular disseminada (CIVD) também pode levar a um aumento isolado de TP, embora seja mais comum observar o prolongamento de ambos, TP e TTPa, simultaneamente. Apesar das reduções dos outros fatores de coagulação, o FVIII pode estar aumentado na CIVD, em razão da liberação de FvW pelas células endoteliais, fato que o torna uma proteína de fase aguda. A coagulação pode ocorrer pela ativação da via do FT e FVII (extrínseca), que além de importante para a conversão de fibrinogênio em fibrina é capaz de ativar outras enzimas e cofatores, FIX e FXI, que retroalimentam a formação do coágulo. A ativação da coagulação também pode ser desencadeada por outras vias devido a expressão de fatores pró-coagulantes como a cisteína-protease ativadora de FX, em pacientes com neoplasias, ou via liberação de moléculas iniciadoras de coagulação em quadros obstétricos. O diagnóstico de CIVD é baseado em achados clínicos e laboratoriais (Figura 2).(17,28,29)
Quando ocorre prolongamento isolado do TTPa
O prolongamento de TTPa de forma isolada aponta para alterações envolvendo a via intrínseca, especialmente FVIII e FIX, podendo estar prolongado também em alterações na via comum e, neste último caso, normalmente associado ao prolongamento de TP. Além disso, o prolongamento isolado de TTPa pode ser observado em amostras contendo heparina, pacientes com presença de AL e pacientes com hematócrito superior a 55% para os quais não foi realizada correção na coleta.(17)
Quando o teste da mistura do TTPa não corrige
A não correção do TTPa após teste da mistura, realizada de imediato ou após incubação, sugere a presença de inibidores. Nestes casos, incubação é particularmente importante devido aos inibidores de FVIII que são anticorpos que necessitam de temperatura adequada para exercer sua ação inibitória. Considerando o histórico do paciente, em casos de eventos trombóticos a presença de AL deve ser investigada.(7,10,18,19,30,31)
Inibidores de FVIII
Pacientes portadores de hemofilia A hereditária podem desenvolver aloanticorpos ao receberem tratamento contendo FVIII exógeno que geralmente são do tipo IgG4. Cerca de 20% a 30% desses pacientes desenvolvem inibidores contra o FVIII. De forma menos comum, há relatos de pacientes sem histórico familiar de sangramentos ou diagnóstico de hemofilia, que desenvolveram autoanticorpos contra FVIII endógeno. Os anticorpos IgA ou IgM também foram descritos em pacientes com hemofilia A associada a doenças hematológicas malignas, puerpério, dentre outras situações, porém também podem ocorrer em pacientes sem doença prévia associada. A presença de inibidores de FVIII pode interferir na atividade de outros fatores da via intrínseca e esse problema pode ser solucionado através da diluição da amostra. Inicialmente é verificado um prolongamento de TTPa, que não corrige após TM (1:1, 1:4 e 4:1). A mistura deve, também, ser incubada durante 2 horas a 37°C, uma vez que os inibidores do FVIII possuem ação lenta (Figura 3). Assim como para outros inibidores, o ensaio de Bethesda é o teste de escolha para detecção de inibidores de FVIII.(8,11,27,31-34)
Recentemente foi aprovado, pelos órgãos competentes, o uso do medicamento emicizumabe, que é projetado para mimetizar a função de cofator de FVIII, reconhecendo os FIX e FX ativados, permitindo a formação do arranjo apropriado para a ativação de FX e assim prevenir sangramentos. Desse modo, o emicizumabe se encontra como uma alternativa aos pacientes que não são responsivos à infusão de FVIII devido a sensibilização prévia e formação de anticorpos anti-FVIII. É importante destacar que o emicizumabe altera os resultados de TTPa e de atividade de FVIII, quando analisados por método coagulométrico. Observa-se uma redução no TTPa e na dosagem de FVIII uma diminuição espúria da atividade. Isso ocorre independentemente do tipo de ativador utilizado no ensaio. A dosagem de FVIII, nestes casos, deverá ser realizada por método cromogênico e com reagentes de origem bovina.(35-38)
Inibidores de FIX
Da mesma forma que surgem anticorpos contra FVIII, anticorpos contra FIX também são encontrados e de maneira mais frequente em pacientes com doenças autoimunes, malignas e com mutações genéticas do tipo grandes deleções, em associação com casos de alergias e reações anafiláticas graves. Contudo, o surgimento de inibidores contra o FIX possui incidência de 5 a 10 vezes inferior à de inibidores de FVIII.(12,32,33)
Inibidores de FXI, e FXIII
Casos de inibidores de FXI são raros e já foram descritos em pacientes após infecções respiratórias virais, em pacientes com lúpus eritematoso sistêmico (LES) e após a infusão de hemocomponentes. Já o desenvolvimento de anticorpos (inibidores) contra FXIII (deficiência adquirida) é raro, na maioria da vezes idiopático, ocorrendo com maior frequência em idosos, gestantes, politransfundidos, pacientes com neoplasias hematológicas, tumores sólidos, doenças autoimunes e em uso de determinados medicamentos (penicilina e amiodarona). As manifestações clínicas podem ser graves e fatais. Tais inibidores podem ser neutralizantes de FXIII e não neutralizantes de FXIII.(39)
Presença de anticoagulantes
A heparina é um potente anticoagulante amplamente utilizado em pacientes hospitalizados para prevenir e tratar trombose e a sua administração deve ser acompanhada de controle laboratorial pelo TTPa (heparina de alto peso molecular – não fracionada) e pelo teste anti-Xa (heparina de baixo peso molecular – fracionada). A heparina é capaz de se ligar à antitrombina e fazer com que esta exponha seu sítio ativo, inibindo a ação do FII e FXa e impedindo a formação do coágulo. No entanto, a heparina pode ser um interferente laboratorial indesejado quando sua presença na amostra é resultante de contaminação durante a coleta. A presença de heparina, a depender da quantidade, torna a amostra incoagulável, de modo que não seja possível a realização dos exames de triagem. Existem aditivos presentes em reagentes de TTPa e de teste de veneno da víbora de Russell diluído (dRVVT) que neutralizam a heparina contaminante da amostra. Os principais aditivos são heparinases, protamina e polibreno. Neutralizantes de inibidores diretos de trombina (IDT) ou inibidores diretos do fator Xa também estão disponíveis, apesar de ainda não serem utilizados em larga escala.(18,40,41)
A detecção de contaminação da amostra por heparina e IDT pode ser realizada pelo TT e pelo anti-Xa. O TT permite verificar a conversão de fibrinogênio em fibrina, avaliando diretamente o fibrinogênio funcional. Desta forma, é especialmente útil em casos de TTPa prolongado, pois está normalmente prolongado nos casos em que o TTPa prolongado ocorre pela presença de heparina, dabigatrana ou deficiência de fibrinogênio. Um TM do TT sem correção sugere a presença de heparina ou dabigatrana, ao passo que a correção sugere deficiência de fibrinogênio. Existem diferentes possibilidades para explicar os TC prolongados em cada situação e o resultado dos TM ajudam a direcionar quais testes adicionais podem ser úteis.(3) Já o anti-Xa é realizado por método cromogênico, onde a antitrombina III e o FXa se encontram em excesso na reação e a taxa de inibição do FXa pelo complexo heparina-antitrombina III é diretamente proporcional aos níveis de heparina, de modo que a atividade de FXa remanescente medida em substrato cromogênico seja inversamente proporcional aos níveis de heparina. Entretanto, em indivíduos com deficiência de antitrombina III do tipo II (deficiência qualitativa) pode não ser possível a detecção de atividade anti-Xa. O paciente pode apresentar resultados de atividade indetectável, e nessa situação os testes da mistura com PPN podem ser realizados a fim de possibilitar o estabelecimento do valor de atividade, através de um ensaio cromogênico baseado em FXa. (Figura 4).(8,17,42,43)
Presença de anticoagulante lúpico
A presença de AL, que é um anticorpo com afinidade por fosfolipídios, predispõe ao risco de trombose e complicações gestacionais, podendo levar a abortos de repetição e nascimentos prematuros. Para fins diagnósticos são avaliados três desses anticorpos: anti-β2GPI, anticorpos anticardiolipina (aCL) e AL. O AL é avaliado por sua capacidade de prolongar testes de coagulação in vitro. Existem três principais diretrizes que orientam a respeito da detecção de AL: a elaborada pela ISTH em 2009, atualizada em 2020; a elaborada pelo CLSI em 2014 e a do BCSH de 2012. Os três documentos possuem em comum orientações a respeito do uso dos TM na detecção de AL. Quando o dRVVT e/ou TTPa se encontram prolongados, é recomendado prosseguir com os TM na proporção 1:1, em que a não correção indica que há presença de inibidor de coagulação que pode ou não ser o AL, a depender se há ou não correção na fase confirmatória com utilização de reagentes com elevado teor de fosfolipídios.(7,18,30,44)
Outros testes podem ser utilizados para a triagem de AL no laboratório de hemostasia como, por exemplo, o tempo de coagulação com kaolin (KCT) e o tempo de protrombina diluído (dTP). O uso do KCT é desencorajado pela diretriz da ISTH devido a sua baixa reprodutibilidade, ao passo que o dTP apresenta bom desempenho quando se utiliza tromboplastina recombinante, que possui alta sensibilidade aos AL. Sabe-se que, apesar de possível, é raro o prolongamento do TP devido a presença de AL, e isso normalmente ocorre porque os reagentes de tromboplastina contêm altas concentrações de fosfolipídios, que anulam os efeitos de AL. Entretanto, quando a tromboplastina é diluída, o AL passa a interferir no tempo de coagulação (TC) do TP. Dessa forma, o dTP pode ser utilizado como triagem para AL e o teste confirmatório correspondente pode ser realizado através da utilização de uma diluição menor de tromboplastina.(11,19,45)
A Figura 5 traz detalhes sobre as diferentes estratégias para pesquisa de AL. Idealmente, a pesquisa de AL deve ser realizada quando o paciente não está fazendo uso de terapia anticoagulante, porém muitas vezes é clinicamente inviável suspender a medicação. A presença de anticoagulantes na amostra do paciente pode ser contornada por meio de estratégias para neutralizar anticoagulantes, da mesma forma como citado para heparina. Anticoagulantes orais diretos (AOD) como os IDT ou inibidores diretos do fator Xa podem ser neutralizados por idarucizumabe, dentre outros. Não há consenso entre as principais diretrizes sobre a recomendação do uso desses neutralizantes. As heparinas exercem efeito sobre os ensaios de AL devido interferirem no TTPa e no dRVVT. Já em plasmas de pacientes em uso de AVK, que apresentem valores de RNI entre 1,5 e 3,0, é possível preparar uma diluição 1:1 de plasma do paciente e PPN, a fim de compensar a ausência de fatores dependentes de vitamina K. Já em relação aos IDT e inibidores diretos do FXa, os efeitos nos ensaios são imprevisíveis.(18,41,44)
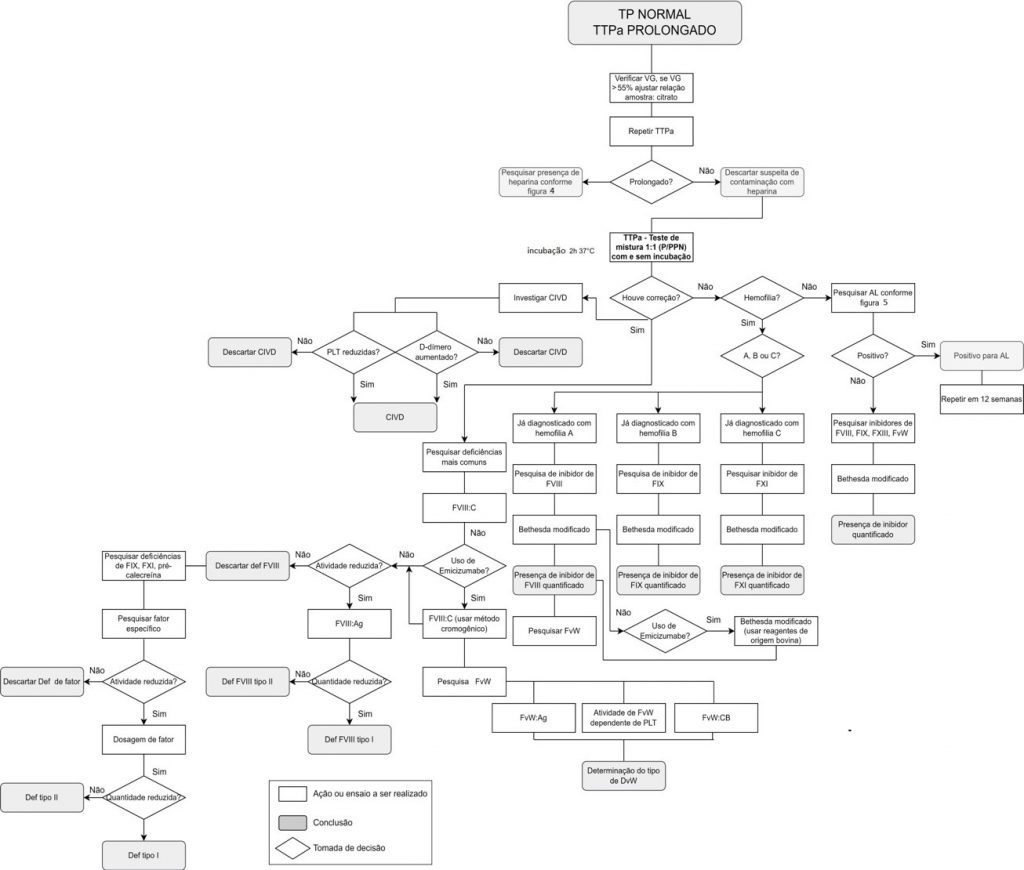
Figura 3
Prolongamento isolado de tempo de tromboplastina parcial ativada (TTPa)
Legenda: P/PPN (relação entre o tempo de coagulação do plasma do paciente e o pool de plasmas normais); def (deficiência); CIVD (coagulação intravascular disseminada); PLT (plaquetas); AL (anticoagulantes lúpicos); VG (volume globular); FVIII:C (pesquisa de atividade de FVIII); FVIII (pesquisa de antígeno FVIII, pesquisa quantitativa); FvW:Ag (teste de fator de von Willebrand antígeno); FvW:CB (teste de ligação do fator de von Willebrand ao colágeno).
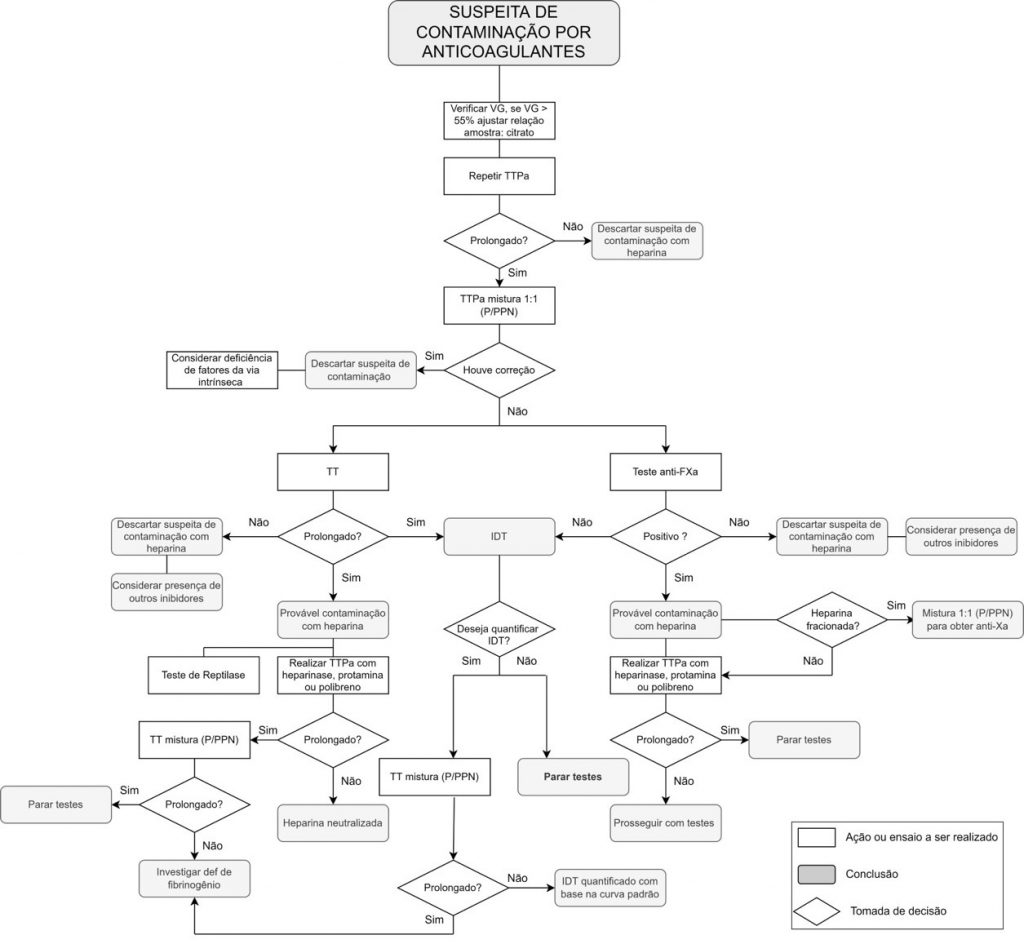
Figura 4
Pesquisa de contaminação por medicamentos anticoagulantes
Legenda: VG (volume globular); P/PPN (relação entre o tempo de coagulação do plasma do paciente e o pool de plasmas normais); IDT (Inibidores Diretos da Trombina).
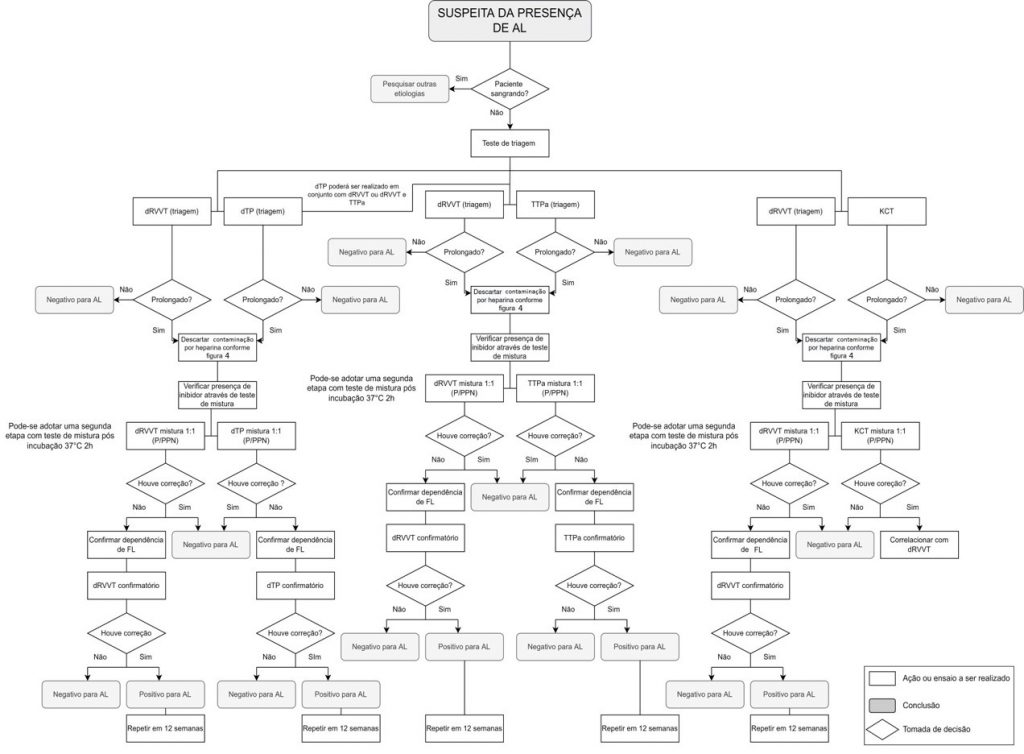
Figura 5
Pesquisa de Anticoagulantes Lúpicos (AL)
Legenda: P/PPN (relação entre o tempo de coagulação do plasma do paciente e o pool de plasmas normais); AL (anticoagulantes lúpicos), FL (fosfolipídio), dTP (teste do tempo de protrombina diluído), dRVVT (teste do veneno da víbora de Russell diluído), KCT (kaolin clotting time – tempo de coagulação pelo caulim)
Quando o teste da mistura do TTPa corrige
Situações já descritas, como CIVD e deficiência de fatores da via intrínseca, com maior frequência a deficiência congênita de FVIII, resultam na correção do TTPa após teste da mistura.
Hemofilia A – Deficiência de FVIII
A hemofilia A é uma doença hereditária de herança ligada ao X, mais frequente em homens, que se caracteriza pela deficiência de FVIII. Acomete cerca de 1:10.000 nascimentos masculinos. O quadro clínico é marcado por hemorragias recorrentes, especialmente hemartroses, e em casos mais graves hemorragias de sistema nervoso central. A doença pode ser classificada de forma grave, moderada ou leve considerando a quantidade de FVIII. A atividade de FVIII inferior a 1% caracteriza hemofilia grave, e desse modo é desejável que os ensaios laboratoriais possuam linearidade em valores inferiores a 1%. A amostra ideal para a pesquisa de FVIII é sangue total, coletado em tubo contendo citrato, no entanto a coleta de sangue de cordão umbilical também pode ser realizada, especialmente em recém-nascidos do sexo masculino. Nestes casos, no entanto, os resultados devem ser interpretados com cautela, visto que durante o parto há ativação da cascata de coagulação e as amostras de cordão tendem a vir parcialmente coaguladas. O FVIII circula complexado ao FvW, de modo que a deficiência de FvW pode resultar em um aumento da depuração de FVIII. Ao diagnóstico se observa TTPa prolongado que corrige após o TM.(8,12,27)
Hemofilia B – Deficiência de FIX
A hemofilia B também é uma doença hereditária ligada ao cromossomo X, que acarreta a deficiência de FIX. Apresenta uma grande variedade de variantes genéticas, o que dificulta a correlação com a clínica. Ao diagnóstico se observa TTPa prolongado que corrige após o TM.(27,39)
Deficiência de FXI, FXII e pré-calecreína
A deficiência de FXI, também chamada de hemofilia C, é uma doença de herança autossômica e incidência rara. A relação entre os níveis de FXI e as manifestações clínicas não é linear. A deficiência de fator FXII é tipicamente autossômica recessiva, com incidência <1:1.000.000, e resulta em prolongamento mais acentuado do TTPa, porém não há manifestações clínicas envolvendo episódios de sangramento, assim como deficiência de pré-calecreína (PC). Ao diagnóstico se observa TTPa prolongado que corrige após o TM em todas essas situações. Contudo, na deficiência de PC, um TTPa prolongado pode diminuir ou até mesmo normalizar caso a amostra seja incubada por tempo suficiente para que a PC diminuída consiga ativar a via intrínseca da coagulação.(27)
Deficiência de fator de von Willebrand
A doença de von Willebrand é uma doença hemorrágica hereditária que afeta cerca de 2% da população e possui herança autossômica irregular. Para estabelecer o diagnóstico é importante que se conheça o histórico familiar e de sangramentos do paciente. O diagnóstico é baseado nos testes de triagem, havendo prolongamento de TTPa, seguido da avaliação de FVIII:C (atividade), FvW:Ag (antígeno) e determinação da função do FvW através de: FvW:RCo (teste cofator de ristocetina) e/ou FvW:GPIbM (teste de atividade de FvW de ligação de GPIb) e FvW:CB (ligação do FvW ao colágeno). Existem inibidores de FvW que se desenvolvem em alguns pacientes com doença de von Willebrand do Tipo 3 e podem ser detectados pela inibição que provocam na atividade do cofator da ristocetina e na atividade do FVIII. Neste último caso, ocorre prolongamento do TTPa e correção do TM do TTPa.(31)
Quando ocorre prolongamentos simultâneos do TTPa e do TP
O prolongamento simultâneo de TTPa e TP aponta para alterações envolvendo a via comum, ou múltiplas vias. Variáveis analíticas e pré-analíticas, como volume de amostra, temperatura de armazenamento da amostra, tempo de armazenamento da amostra antes do processamento e uso de ativadores, devem ser avaliadas, visto que podem levar ao prolongamento dos exames de triagem. Descartadas as possibilidades anteriores, deve-se prosseguir com a investigação de deficiência congênita ou adquirida de fatores da via comum.(9,10,31)
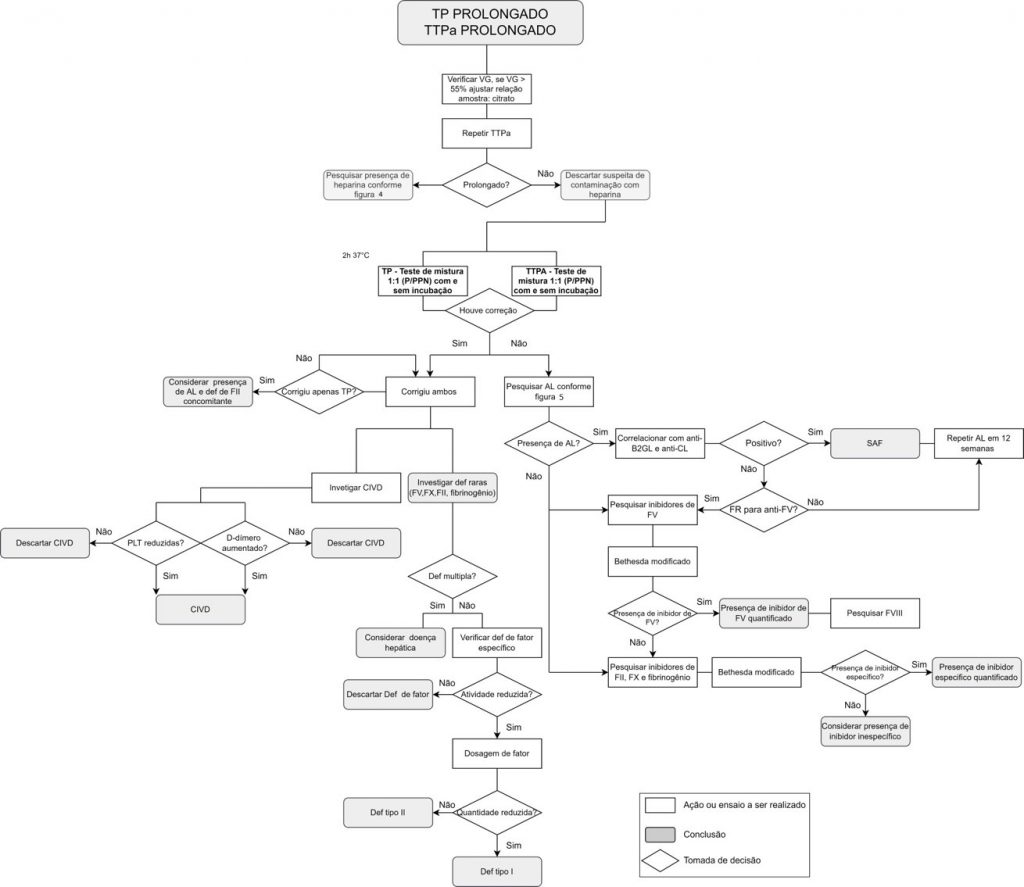
Figura 6
Prolongamentos simultâneos de TTPa e de TP
Legenda: P/PPN (relação entre o tempo de coagulação do plasma do paciente e o tempo do pool de plasmas normais); def (deficiência); CIVD (coagulação intravascular disseminada); PLT (plaquetas); AL (anticoagulantes lúpicos); VG (volume globular); anti-B2GL (anti-beta-2-glicoproteína I); anti-CL (anti-cardiolipina); SAF (síndrome antifosfolipídio); FR (fator de risco).
Quando os testes da mistura do TTPa e do TP não corrigem
A não correção do TP e do TTPa, após a realização dos respectivos TM, direciona a investigação para a presença de inibidores. Investiga-se a presença de inibidores específicos para fibrinogênio, FII, FV e FX, sendo mais comumente verificada a presença de inibidores direcionados ao FV (Figura 6).(27,34)
Inibidores de FV
Inibidores de FV são frequentemente observados em pacientes que possuem fatores de risco como intervenções cirúrgicas, uso de beta-lactâmicos, aminoglicosídeos e histórico de transfusões. Há relatos de casos de desenvolvimento de inibidores contra FV após a utilização de hemostáticos tópicos, como colas cirúrgicas, contendo trombina bovina e vestígios de FV bovino, capazes de estimular o sistema imune. Devido à similaridade estrutural entre FV e FVIII, autoanticorpos formados contra FV podem interferir na atividade de FVIII. O diagnóstico diferencial entre a presença de AL e inibidor de FV é realizado com o auxílio dos TM, onde a não correção de TTPa após a incubação com reagente rico em fosfolipídio indica a presença de inibidor de FV e não de AL. Por outro lado, alguns autoanticorpos anti-FV fogem à regra, de modo que o resultado da pesquisa de AL deve ser correlacionado com a história clínica e sintomatologia, sendo verificado casos de predisposição a trombose em pacientes que possuem AL e sangramentos no caso de inibidores de FV.(34,39,46-48)
Inibidores de FX, FII e fibrinogênio
O FX, quando ativado, catalisa a conversão de protrombina em trombina, e a deficiência adquirida de FX está associada frequentemente à amiloidose sistêmica, que provoca redução de FX na circulação. Há relatos do surgimento de autoanticorpos contra o FX em situações de infecção respiratória e leucemia mielóide aguda, apesar de não ter sido estabelecida relação de causalidade. Inibidores de FII (protrombina) podem ser observados em pacientes com síndrome antifosfolipídio (SAF), LES, artrite reumatoide e em pacientes que receberam preparados de trombina de origem bovina. Parte significativa desses anticorpos não estão associados a sangramentos, todavia episódios hemorrágicos são observados em alguns pacientes. Na SAF existe a possibilidade de desenvolvimento de síndrome de anticoagulante lúpico-hipoprotrombinêmica, que causa predisposição à trombose e a sangramentos. Anticorpos inibidores de fibrinogênio ocorrem de forma rara e são observados em pacientes que receberam reposição de hemocomponentes. Estão associados a episódios graves de sangramento, entretanto há relatos de altos níveis de anticorpos antifibrinogênio não associados a hemorragias em gestantes e puérperas, especialmente em casos de mulheres sensibilizadas devido a diferença de fator Rh entre mãe e recém-nascido e em partos difíceis.(22,32,39,49,50)
Quando os testes da mistura do TTPa e do TP corrigem
Como descrito anteriormente, a CIVD pode levar ao prolongamento de TP e de TTPa, devendo ser investigada. A correção após teste da mistura direciona a pesquisa para deficiência de fatores da via comum. (27)
Deficiência hereditária de FV
A deficiência de FV congênita é uma doença autossômica recessiva, rara, com incidência estimada de 1:1.000.000 e caracterizada por níveis de atividade de FV baixos ou indetectáveis. Pode ser classificada em tipo I e tipo II e os pacientes apresentam uma variedade de manifestações clínicas, desde assintomáticos até epistaxe, hematomas e hemorragias intracranianas. Além de ser encontrado no plasma, o FV pode ser encontrado nos grânulos densos das plaquetas, havendo evidências de que o FV proveniente das plaquetas apresenta atividade pró-coagulante mais pronunciada, quando comparada ao fator plasmático. O papel do FV proveniente das plaquetas ainda não está bem estabelecido, no entanto há grupos de pacientes com deficiência de FV que apresentam manifestações hemorrágicas graves e atividade de FV plaquetário indetectável. Laboratorialmente, a pesquisa de deficiência de FV deve ser realizada com base nos resultados de TTPa e TP prolongados e corrigidos após teste da mistura, seguida de determinação da atividade do FV (Figura 6).(11,27,34,47)
Deficiência hereditária de FX
A deficiência de FX é uma doença com prevalência aproximada de 1:1.000.000 e possui caráter autossômico recessivo. Os episódios de sangramento tendem a ocorrer em pacientes com níveis de FX inferiores a 10%. Os níveis fisiológicos de FX estão elevados durante a gestação em mais de 150% e no período pós-parto. Laboratorialmente, a pesquisa de deficiência de FX deve ser realizada com base nos resultados de TTPa e TP prolongados e corrigidos após teste da mistura, seguida de determinação da atividade do FX.(25,51)
Deficiência hereditária de FII
A prevalência da deficiência hereditária de FII é de aproximadamente 1:2.000.000, sendo extremamente rara. Pode ser observada hipoprotrombinemia, ou deficiência do tipo I, na qual o diagnóstico é realizado como base nos baixos níveis de atividade do FII. A disprotrombinemia, ou deficiência do tipo II, resulta de uma síntese quase normal de protrombina circulante, porém a proteína de alguma forma é disfuncional, com atividade reduzida, mas a quantificação de antígeno se encontra dentro dos valores esperados. Em pacientes com níveis de FII inferiores a 5% no plasma, as manifestações clínicas são sangramentos graves. Entretanto, de forma paradoxal, há relatos na literatura de casos envolvendo pacientes com deficiência conhecida de FII e desenvolvimento de trombose venosa profunda. Laboratorialmente, a pesquisa de deficiência de FII deve ser realizada com base nos resultados de TTPa e TP prolongados e corrigidos após teste da mistura, seguida de determinação da atividade do FII.(52)
Deficiência de fibrinogênio
A afibrinogenemia, doença autossômica dominante, é caracterizada pela total ausência de produção de fibrinogênio, normalmente diagnosticada ao nascimento, devido ao sangramento de cordão umbilical. Nesta condição todos os testes dependentes da formação de fibrina estão prolongados e o fibrinogênio se encontra indetectável, tanto em teste funcional (Claus), quanto em ensaios antigênicos. As deficiências de fibrinogênio também podem ser classificadas em hipofibrinogenemia, na qual há produção inferior aos níveis normais e disfibrinogenemia, na qual a produção da proteína se mantém normal em níveis quantitativos, porém com alterações qualitativas.
Na hipofibrinogenemia, o TT é um ensaio muito sensível, e pode ser confirmado por um tempo de reptilase anormal e níveis de fibrinogênio inferiores ao intervalo de referência. Pode ser decorrente de doença congênita, porém deve-se investigar função hepática. O TM do TT pode ser útil no diagnóstico de disfibrinogenemia, visto que a quantificação de fibrinogênio por método imunoenzimático não avalia a quantidade de fibrinogênio funcional. No TM do TT já foi observada a correção total após mistura 1:5 (P/PPN), no entanto já foi possível observar correção de 50% dos pacientes em mistura 1:4.
A não correção indica a potencial presença de inibidor. Na disfibrinogenemia, geralmente há uma discrepância entre os testes dependentes da formação de fibrina e o fibrinogênio medido antigenicamente. Entre as vantagens do teste tempo de reptilase, destaca-se a sua insensibilidade à presença de heparina não fracionada e deficiência de FXIII, de modo que o ensaio pode ser útil na detecção de hipofibrinogenemia ou disfibrinogenemia em amostras de pacientes contendo heparina.(8,27,53,54)
CONSIDERAÇÕES FINAIS
Os TM são normalmente indicados na investigação de prolongamentos do TTPa ou TP, sendo também utilizados na investigação de anticoagulantes lúpicos. Não são indicados quando o tempo de coagulação dos testes de triagem encontram-se dentro dos valores de referência e até mesmo levemente acima desse intervalo. Antes da realização de um TM, devem ser obtidas informações sobre o histórico do paciente, incluindo uso medicamentos, assim como se a amostra foi coletada adequadamente.(13)
O papel fundamental dos TM pode ser simplesmente expresso como um método para distinguir deficiência de fator de coagulação de um inibidor. O principal desafio para o laboratório é estabelecer critérios confiáveis que maximizem as chances de interpretação correta do resultado do TM. Existem diversas fórmulas que os laboratórios podem escolher para a interpretação dos TM, ressaltando-se que é uma boa prática de laboratório o estabelecimento de valores de corte locais considerando o analisador, PPN e tipos de reagentes em uso.(52,55)
Na aplicação clínica e laboratorial dos TM podemos observar três cenários básicos: apenas TP prolongado, apenas TTPa prolongado ou TP e TTPa prolongados, em que os TM são realizados apenas para o teste em que ocorreu prolongamento. Testes adicionais podem ser recomendados dependendo do contexto clínico e a critério médico. Os vários tipos de situações envolvendo os testes de triagem em hemostasia, assim como os TM e estudos adicionais, são mostrados na Tabela 1.
A pesquisa de alterações da hemostasia no laboratório clínico deve ser realizada de forma cuidadosa e sistemática, sendo fundamental o conhecimento aprofundado do tema por parte dos analistas clínicos e dos médicos, sob o risco de gerar prejuízos no diagnóstico e acompanhamento dos pacientes. A aplicação, o desempenho e a interpretação inadequadas dos testes de triagem e dos TM em hemostasia podem levar a erros diagnósticos na classificação da causa da coagulação anormal, podendo acarretar tratamento incorreto dos pacientes. Cabe aqui ressaltar que decisões clínicas não devem ser guiadas por resultados isolados de testes de triagem e dos TM e sim no contexto clínico-laboratorial.
Por fim, conclui-se que os TM oferecem benefícios substanciais, pois ajudam de fato a orientar a investigação para diferenciar deficiência de fatores de coagulação de presença de inibidor, reduzindo custos e tempo e maximizando a eficiência diagnóstica para os pacientes.
Tabela 1
Comportamento dos testes de coagulação em diversas situações clínicas
| Situações | Teste de coagulação | ||||||
| TP | TTPa | TT | TP mix | TTPa mix | TT mix | Testes adicionais para confirmação | |
| Deficiências hereditárias de fatores de coagulação | |||||||
| FVIII / FIX / FXI / FXII / PC / CAPM | Normal | Prolongado | Normal | – | Corrige | – | Dosagem de
FVIII, FIX, FXI, FXII, PC e CAPM |
| FII / FV / FX | Prolongado | Prolongado | Normal | Corrige | Corrige | – | Dosagem de FII, FV e FX |
| FVII | Prolongado | Normal | Normal | Corrige | – | – | Dosagem de FVII |
| Afibrinogenemia, disfibrinogenemia ou hipofibrinogenemia | Prolongado | Prolongado | Prolongado | Corrige | Corrige | Corrige | Dosagem de fibrinogênio |
| FXIII | Normal | Normal | Normal | – | – | – | Dosagem de FXIII |
| Doença de von Willebrand com FVIII baixo | Normal | Prolongado | Normal | – | Corrige | – | Dosagem de FVIII e FvW |
| Deficiências adquiridas de fatores de coagulação | |||||||
| Doenças hepáticas | Prolongado | Prolongado | Normal ou Prolongado | Corrige | Corrige | Corrige | Dosagem de fatores e de enzimas hepáticas |
| Deficiência de vitamina K | Prolongado | Prolongado | Normal | Corrige | Corrige | – | Dosagem de FII, FVII, FIX e FX |
| CIVD | Prolongado | Prolongado | Prolongado | Corrige | Corrige | Corrige | Dosagem de fatores, dímero D, fibrinogênio e plaquetas |
| Amiloidose sistêmica com perda de FX | Prolongado | Prolongado | Normal | Corrige | Corrige | – | Dosagem de FX |
| Síndrome nefrótica com perda de FXII | Normal | Prolongado | – | – | Corrige | – | Dosagem de FXII |
| FVIII diminuído secundariamente a inibidor de FvW | Normal | Prolongado | Normal | – | Corrige | – | Dosagem de FVIII e FvW |
| Inibidores de fatores de coagulação do tipo anticorpos | |||||||
| Anticoagulante lúpico | Normal ou raramente aumentado | Prolongado | Normal | Variável | Não corrige | – | dRVVT |
| Inibidor de FVIII | Normal | Prolongado | Normal | – | Correção imediata variável e não correção tardia após incubação | – | Dosagem de FVIII e de inibidor de FVIII pelo método de Bethesda modificado |
| Inibidor de FIX, FXI ou FXIII | Normal | Prolongado | Normal | – | Não corrige | – | Dosagem de FIX, FXI ou FXIII e de inibidor de FIX ou FXI pelo método de Bethesda modificado ou de FXIII por teste da mistura do tipo neutralizante ou por imunoensaio de ligação |
| Inibidor de FVII | Prolongado | Normal | Normal | Não corrige | – | – | Dosagem de FVII e de inibidor de FVII pelo método de Bethesda modificado |
| Inibidor de FII, FV ou FX | Prolongado | Prolongado | Normal | Não corrige | Não corrige | – | Dosagem de FII, FV ou FX e de inibidor de FII, FV ou FX pelo método de Bethesda modificado |
| Agentes terapêuticos | |||||||
| Heparina não fracionada
(níveis terapêuticos) |
Normal desde que reagente contenha neutralizador de heparina | Prolongado | Prolongado | Corrige | Não corrige | Variável | Anti-Xa dentro dos níveis terapêuticos. TT prolongado com não correção no TT mix é sugestivo, mas também pode ocorrer com o uso de inibidores diretos da trombina |
| Heparina não fracionada
(acima dos níveis terapêuticos) |
Prolongado | Prolongado | Prolongado | Variável | Não corrige | Não corrige | Anti-Xa acima dos níveis terapêuticos. TT prolongado com não correção no TT mix é sugestivo, mas também pode ocorrer com o uso de inibidores diretos da trombina |
| Heparina de baixo peso molecular | Normal | Normal ou prolongado | Normal | – | – | – | Monitoramento pelo teste anti-Xa |
| Antagonistas da vitamina K
(início da terapia onde vit. K encontra-se levemente diminuída) |
Prolongado | Normal, raramente prolongado | Normal | Corrige | Corrige | – | FII, FVII, FIX e FX estarão baixos |
| Antagonistas da vitamina K
(longos períodos em níveis terapêuticos, vit. K encontra-se bastante diminuída) |
Prolongado | Prolongado | Normal | Corrige | Corrige | – | FII, FVII, FIX e FX estarão baixos |
| Argatrobana e Dabigatrana
(inibidores diretos da trombina) |
Normal ou prolongado | Prolongado, raramente normal | Prolongado | Não corrige | Não corrige | Normalmente não corrige | Dosagem de inibidor direto da trombina |
| Apixabana, Edoxabana e Rivaroxabana (inibidores de FXa) | Prolongado | Normal ou prolongado | Normal | Não corrige | Normalmente não corrige | – | Dosagem de inibidor de FXa |
Legenda: TTPa, tempo de tromboplastina parcial ativada; PC, pré-calicreína; CAPM, cininogênio de alto peso molecular; TP, tempo de protrombina; TT, tempo de trombina; FvW, fator de von Willebrand; CIVD, coagulação intravascular disseminada; FII, fator II da coagulação; FV, fator V da coagulação; FVII, fator VII da coagulação; FVIII, fator VIII da coagulação; FIX, fator IX da coagulação; FX, fator X da coagulação; FXI, fator XI da coagulação; FXII, fator XII da coagulação; Xa, X ativado; mix, teste da mistura.
REFERÊNCIAS
- Kriz N, Rinder CS, Rinder HM. Physiology of Hemostasis: with relevance to current and future laboratory testing. Clin Lab Med. 2009;29(2):159-174.
- Bonar RA, Lippi G, Favaloro EJ. Overview of hemostasis and thrombosis and contribution of laboratory testing to diagnosis and management of hemostasis and thrombosis disorders. In: Favaloro EJ, Lippi G (Eds). Hemostasis and Thrombosis: Methods and Protocols. New York: Springer Nature, 2017. p. 3-27.
- Favaloro EJ. Coagulation mixing studies: utility, algorithmic strategies and limitations for lupus anticoagulant testing or follow up of abnormal coagulation tests. Am J Hematol. 2020;95(1):117-128.
- Tripodi A, Lippi G, Plebani M. How to report results of prothrombin and activated partial thromboplastin times. Clin Chem Lab Med. 2016;54(2):215-222.
- Dorgalaleh A, Favaloro EJ, Bahraini M, Rad F. Standardization of prothrombin time/international normalized ratio (PT/INR). Int J Lab Hematol. 2021;43(1):21-28.
- Chee Y. Coagulation. J R Coll Physicians Edinb. 2014;44(1):42-45.
- Devreese KMJ, Groot PG, Laat B, Erkan D, Favaloro EJ, Mackie I, et al. Guidance from the scientific and standardization committee for lupus anticoagulant/antiphospholipid antibodies of the International Society on Thrombosis and Haemostasis: update of the guidelines for lupus anticoagulant detection and interpretation. J Thromb Haemost. 2020;18(11):2828-2839.
- BRASIL. Ministério da Saúde. Manual de diagnóstico laboratorial das coagulopatias hereditárias e plaquetopatias. Brasília: Editora MS 2016, 140p.
- Benzon HT, Park M, McCarthy RJ, Kendall MC, Lindholm PF. Mixing studies in patients with prolonged activated partial thromboplastin time or prothrombin time. Anesth Analg. 2019;128(6):1089-1096.
- Kamal AH, Tefferi A, Pruthi RK. How to interpret and pursue an abnormal prothrombin time, activated partial thromboplastin time, and bleeding time in adults. Mayo Clin Proc. 2007;82(7):864-873.
- Kershaw G, Orellana D. Mixing tests: diagnostic aides in the investigation of prolonged prothrombin times and activated partial thromboplastin times. Semin Thromb Hemost. 2013;39(3):283-290.
- Kershaw G, Favaloro EJ. Laboratory identification of factor inhibitors: an update. Pathology. 2012;44(4):293-302.
- Adcock DM, Moore GW, Montalvão SL, Kershaw G, Gosselin RC. Activated partial thromboplastin time and prothrombin time mixing studies: current state of the art. Semin Thromb Hemost. 2022 Sep 2. doi: 10.1055/s-0042-1756196. Epub ahead of print.
- Rosner E, Pauzner R, Lusky A, Modan M, Many A. Detection and quantitative evaluation of lupus circulating anticoagulant activity. Thromb Haemost. 1987;57(2):144-147.
- Chang SH, Tillema V, Scherr D. A “percent correction” formula for evaluation of mixing studies. Am J Clin Pathol. 2002;117(1):62-73.
- Kershaw G, Jayakodi D, Dunkley S. Laboratory identification of factor inhibitors: the perspective of a large tertiary hemophilia center. Semin Thromb Hemost. 2009 Nov; 35(08):760-8.
- Kottke-Marchant K. Algorithmic approaches to hemostasis testing. Semin Thromb Hemost. 2014;40(2):195-204.
- Pengo V, Tripodi A, Reber G, Rand JH, Ortel TL, Galli M, et al. Update of the guidelines for lupus anticoagulant detection. J Thromb Haemost. 2009 Oct;7(10):1737–40.
- Keeling D, Mackie I, Moore GW, Greer IA, Greaves M, British Committee for Standards in Haematology. Guidelines on the investigation and management of antiphospholipid syndrome. Br J Haematol. 2012 Apr;157(1):47-58.
- Clinical and Laboratory Standards Institute (2005). Procedures for validation of INR and local calibration of PT/INR systems; approved guideline. H54-A, vol. 25, No. 23. Wayne, PA: Clinical and Laboratory Standards Institute.
- Laffan MA, Manning RA. Investigation of haemostasis. In: Bain BJ, Bates I, Laffan MA, Lewis SM. Dacie and Lewis Practical Haematology. 12th ed. London: Elsevier Limited, 2017. p. 366-409.
- Collins PW, Chalmers E, Hart DP, Liesner R, Rangarajan S, Talks K, et al. Diagnosis and treatment of factor VIII and IX inhibitors in congenital haemophilia. Br J Haematol. 2013;160(2):153-170.
- Sevenet PO, Kaczor DA, Depasse F. Factor VII deficiency: from basics to clinical laboratory diagnosis and patient management. Clin Appl Thromb. 2017;23(7):703-710.
- Mariani G, Bernardi F. Factor VII deficiency. Semin Thromb Hemost. 2009;35(4):400-406.
- Kamikubo Y, Mendolicchio GL, Zampolli A, Marchese P, Rothmeier AS, Orje JN, et al. Selective factor VIII activation by the tissue factor–factor VIIa–factor Xa complex. Blood. 2017;130(14):1661-1670.
- Bidot CJ, JY W, Horstman LL, Huisheng H, Jimenez JJ, Yaniz M, et al. Factor VII/VIIa: a new antigen in the anti-phospholipid antibody syndrome: Anti-FVII/VIIa in APS. Br J Haematol. 2003;120(4):618-626.
- Wagenman BL, Townsend KT, Mathew P, Crookston KP. The aboratory approach to inherited and acquired coagulation factor deficiencies. Clin Lab Med. 2009;29(2):229-252.
- Levi M. Pathogenesis and management of peripartum coagulopathic calamities (disseminated intravascular coagulation and amniotic fluid embolism). Thromb Res. 2013;131:S32-34.
- Gando S, Levi M, Toh CH. Disseminated intravascular coagulation. Nat Rev Dis Primer. 2016;2(2):16037.
- Molhoek J, de Groot P, Urbanus R. The lupus anticoagulant paradox. Semin Thromb Hemost. 2018;44(5):445-452.
- Levy JH, Szlam F, Wolberg AS, Winkler A. Clinical use of the activated partial thromboplastin time and prothrombin time for screening. Clin Lab Med. 2014;34(3):453-477.
- Franchini M, Veneri D. Acquired coagulation inhibitor-associated bleeding disorders: an update. Hematology. 2005;10(6):443-449.
- Franchini M, Lippi G, Favaloro E. Acquired inhibitors of coagulation factors: part II. Semin Thromb Hemost. 2012;38(5):447-453.
- Franchini M, Castaman G, Coppola A, Santoro C, Zanon E, Di Minno G, et al. Acquired inhibitors of clotting factors: AICE recommendations for diagnosis and management. Blood Transfus. 2015;13(3):498-513.
- Kitazawa T, Esaki K, Tachibana T, Ishii S, Soeda T, Muto A, et al. Factor VIIIa-mimetic cofactor activity of a bispecific antibody to factors IX/IXa and X/Xa, emicizumab, depends on its ability to bridge the antigens. Thromb Haemost. 2017;117(7):1348-1357.
- Tiede A, Kemkes-Matthes B, Knöbl P. Should emicizumab be used in patients with acquired hemophilia A? J Thromb Haemost. 2021;19(3):637-644.
- Rodriguez EC, Valentino LA. Emicizumab: review of the literature and critical appraisal. Haemophilia. 2019;24(1):11-20.
- Jenkins PV, Bowyer A, Burgess C, Gray E, Kitchen S, Murphy P, et al. Laboratory coagulation tests and emicizumab treatment. A United Kingdom Haemophilia Centre Doctors’ Organisation guideline. Haemophilia. 2020;26(1):151-155.
- Cugno M, Gualtierotti R, Tedeschi A, Meroni PL. Autoantibodies to coagulation factors: from pathophysiology to diagnosis and therapy. Autoimmun Rev. 2014;13(1):40-48.
- Winkler AM, Sheppard CA, Fantz CR. Laboratory monitoring of heparin: challenges and opportunities. Lab Med. 2007;38(8):499-502.
- Ozawa T, Mammen EF. LMW heparin (anti-Xa) assays for clinical monitoring and pharmacokinetic studies on the automated coagulation laboratory (ACL). Thromb Res. 1992;66(4):287-298.
- Favaloro EJ, Pasalic L. Lupus anticoagulant testing during anticoagulation, including direct oral anticoagulants. Res Pract Thromb Haemost. 2022;6(2):e12676.
- Bahabri A, Moffat KA, Carlino SA, Chan AKC, Bhatt MD. Mixing study to diagnose heparin‐resistance caused by functional antithrombin deficiency. Int J Lab Hematol. 2022;44(5):e227-e229.
- Moore GW. Commonalities and contrasts in recent guidelines for lupus anticoagulant detection. Int J Lab Hematol. 2014;36(3):364-373.
- Liestøl S, Jacobsen EM, Wisløff F. Dilute prothrombin time-based lupus ratio test. Integrated LA testing with recombinant tissue thromboplastin. Thromb Res. 2002 Jan 15;105(2):177-82.
- Leus B, Devreese K, Van den Bossche J, Malfait R. Factor V inhibitor: case report. Blood Coagul Fibrinolysis. 2006;17(7):585-587.
- Thalji N, Camire R. Parahemophilia: new Insights into Factor V deficiency. Semin Thromb Hemost. 2013;39(6):607-612.
- Olson NJ, Ornstein DL. Factor V inhibitors: a diagnostic and therapeutic challenge. Arch Pathol Lab Med. 2017;141(12):1728-1731.
- Spada ARL, Skålhegg BS, Henderson R, Schmer G, Pierce R, Chandler W. Fatal hemorrhage in a patient with an acquired inhibitor of human thrombin. N Engl J Med. 1995;333(8):494-497.
- Guddati AK, Kuter DJ. Rituximab therapy for factor II inhibitor in a patient with antiphospholipid antibody syndrome. Blood Coagul Fibrinolysis. 2014;25(3):289-291.
- Hellgren M. Hemostasis during normal pregnancy and puerperium. Semin Thromb Hemost. 2003;29(2):125-130.
- Watson H, Perez A, Ayesu K, Musa F, Sarriera J, Madruga M, et al. Inherited factor II deficiency with paradoxical hypercoagulability: a case report. Blood Coagul Fibrinolysis. 2018;29(2):223-226.
- Chandler JB, Siddon AJ, Bahel P, Torres R, Rinder HM, Tormey CA. Modified approach to fibrinogen replacement in the setting of dysfibrinogenaemia. J Clin Pathol. 2019;72(2):177-180.
- Miesbach W, Schenk J, Alesci S, Lindhoff-Last E. Comparison of the fibrinogen Clauss assay and the fibrinogen PT derived method in patients with dysfibrinogenemia. Thromb Res. 2010;126(6):e428-433.
- Falay M, Senes M, Yücel D, Turhan T, Dagdaş S, Pekin M, et al. What should be the laboratory approach against isolated prolongation of a activated partial thromboplastin time? J Clin Lab Anal. 2018;32(6):e22415.