Feocromocitoma: relato de caso
Pheochromocytoma: case report
Jaqueline Antunes de Ramos1
Rúbia Graziela Ramos Pedrelli1
Magda Helena Soratto Heitich Ferrazza2
Gabriela Borgmann3
Katherine Plautz3
1Farmacêutica. Sociedade Educacional de Santa Catarina (UniSociesc). Jaguará do Sul-SC, Brasil.
2Mestre. Professora de Hematologia e Microbiologia. Sociedade Educacional de Santa Catarina (UniSociesc). Jaguará do Sul-SC, Brasil.
3Graduanda em Biomedicina – Sociedade Educacional de Santa Catarina (UniSociesc). Jaguará do Sul-SC, Brasil.
Instituição: Sociedade Educacional de Santa Catarina (UniSociesc). Jaguará do Sul-SC, Brasil.
Recebido em 18/02/2020
Aprovado em 05/10/2020
DOI: 10.21877/2448-3877.202100957
INTRODUção
Feocromocitomas são tumores que se originam das células cromafins e podem se desenvolver na medula suprarrenal. Esses tumores secretam grande quantidade de catecolaminas, o que leva a um conjunto de efeitos metabólicos, sendo a hipertensão arterial refratária a manifestação mais frequente.(1) A incidência anual do feocromocitoma é de dois a oito novos casos por um milhão de habitantes e afeta ambos os sexos com pico de incidência entre a 4a e 5a décadas de vida. Cerca de 90% dos tumores são benignos e unilaterais. E atualmente 20% a 30% são bilaterais e estão relacionados à herança familiar.(2) A ocorrência de malignidade do feocromocitoma varia entre 3% a 36%, ou seja, um diagnóstico preciso aumenta a taxa de sobrevida.(3)
As manifestações clínicas são promovidas pela hipersecreção das catecolaminas, que propiciam a hipertensão, cefaleia, sudorese e taquicardia.(4) Além da elevação da pressão arterial, o feocromocitoma pode causar outras complicações como: hemorragias retinianas, nefropatia, infarto agudo do miocárdio, (resultante de miocardite, ou de vasoespasmo de artéria coronária) embolia pulmonar, acidente vascular encefálico por infarto cerebral, hemorragia intracraniana ou embolia.(5)
O feocromocitoma é considerado uma doença genética autossômica dominante, conhecido como uma mutação germinativa que ocorre no proto-oncogene Rearranged during transfection (RET) do cromossomo 10q 11.2 e causa a síndrome Neoplasia Endócrina Múltipla tipo 2 (NEM 2A). O gene RET é uma proteína que codifica o receptor transmembranar tirosina quinase, responsável pela regulação celular. O avanço dos exames genéticos têm possibilitado o diagnóstico específico para essa patologia.(6)
Na síndrome (NEM 2A) os feocromocitomas ocorrem em associação com carcinoma medular da tireoide em 59% dos casos.(7) Os feocromocitomas sintetizam e armazenam catecolaminas e seus metabólitos, sendo seu diagnóstico baseado nos exames de imagens e na avaliação do excesso dessas catecolaminas por meio dos exames bioquímicos. Os níveis elevados de catecolaminas plasmáticas e urinárias e seus metabólitos constituem a base para o estabelecimento do diagnóstico.
O estudo relata o caso de um paciente com quadro de crise adrenérgica, sendo diagnosticado feocromocitoma.
Relato de caso
Paciente M.H, 47 anos, masculino, branco, casado, torneiro mecânico, natural e procedente de Corupá, Santa Catarina. Possui histórico de diversas internações anteriores ao ano de 2004 por crise hipertensiva, mas sem diagnóstico conclusivo. O histórico familiar confirma hipertensão arterial sistêmica (HAS), a mãe falecida há três anos apresentava um quadro de hipertensão e litíase renal. Radiografia de abdômen da mãe evidenciou imagem suspeita (nódulo hipodenso) localizada em cima dos rins, porém não foi investigada. Um dos irmãos retrata distúrbios da tireoide, que após rastreamento genético familiar confirmou-se o diagnóstico de Carcinoma Medular de Tireoide (CMT).
Em abril de 2004, o paciente M.H. é encaminhado ao serviço de urgência com crise hipertensiva, taquicardia, palidez e cefaleia lobocraniana generalizada de forte intensidade. Não fazia uso de medicamentos. Negou tabagismo e etilismo social, relatou litíase renal desde 1994.
Evoluiu com piora da pressão arterial, no exame clínico os níveis eram de 300 mmHg x 180 mmHg. A frequência cardíaca variava em torno de 94 a 120 batimentos por minuto. Posteriormente foi internado na Unidade de Terapia Intensiva (UTI) para investigação do quadro clínico. Foram solicitados exames laboratoriais e de imagens.
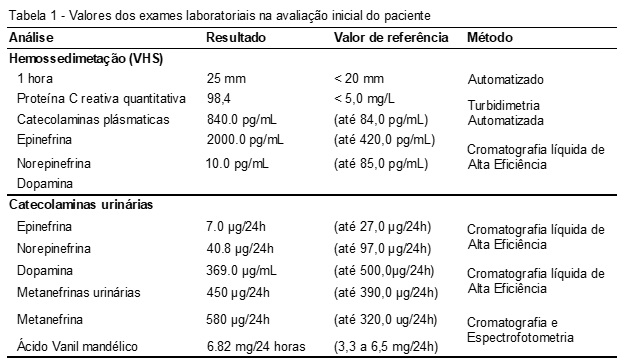
Os exames laboratoriais: hemograma, glicose, colesterol total e frações, ureia, creatinina, sódio, potássio, desidrogenase láctica, bilirrubinas total e frações, exame do líquido cefalorraquidiano (LCR), alanina aminotransferase (ALT), aspartato aminotransferase (AST), fosfatase alcalina, hepatites A, B e C, aldosterona, clearence de creatinina, proteinúria e renina permaneceram dentro dos valores de referência. Já os testes descritos na Tabela 1 estavam elevados comparados aos valores de referência.
Na ressonância magnética (RM) foram encontrados: processo expansivo nodular, medindo aproximadamente 4 cm no maior diâmetro transverso e 4 cm no maior diâmetro longitudinal, bem delimitada, com sinal heterogêneo, predominantemente hiperintenso nas sequências ponderadas em T2 e hipointenso nas sequências ponderadas em T1, com discreto realce heterogêneo após a injeção endovenosa de contraste paramagnético observado na topografia da glândula adrenal direita.
Outra lesão expansiva, medindo aproximadamente 5 cm no maior diâmetro transversal e 4 cm no maior diâmetro longitudinal, com características semelhantes à lesão descrita anteriormente, é observada na topografia da glândula adrenal esquerda. O estudo por RM do abdome superior apresentou lesões nodulares na topografia de glândulas adrenais, sendo considerada a possibilidade de feocromocitoma bilateral com núcleos pouco anisomórficos e o citoplasma denso. Devido às grandes proporções da massa, optou-se pela adrenalectomia bilateral.
O anatomopatológico das glândulas suprarrenais direitas e esquerdas evidenciaram neoplasias compatíveis com feocromocitoma. O estudo imunohistoquímico pela técnica da Avidina-biotina peroxidase e/ou fosfatase alcalina ABC (Cromogranina A, Proteína S-100 e Sinaptofisina positivas) confirmaram o diagnóstico de feocromocitoma.
Após diagnóstico conclusivo, foram realizados exames de imagens da região cervical por meio de ultrassonografia (USG) e cintilografia com sestamibi das glândulas tireoide e paratireoides. As imagens foram obtidas 15 minutos, duas horas e cinco horas após administração endovenosa de 99 Tc-MIBI, na incidência anterior da região cervical e torácica e OAD 45° e OAE 45°. Concluiu-se bócio tireoideano multinodular e ausência de captação em paratireoides detectável pelo método. Em seguida foi encaminhado para tratamento cirúrgico: tireoidectomia total e paratireoidectomia, e avaliação genética por quadro típico da Síndrome NEM tipo 2A.
O laudo do exame histopatológico da tireoide confirma carcinoma medular medindo 2,6 cm x 1,8 cm no lobo direito e 1,5 cm x1,0 cm x1,0 cm e 0,7 cm x 0,6 cm x 0,6 cm no lobo esquerdo, não encapsulado, com extensão para tecidos moles extratireoideanos. Ausência de infiltração angiolinfática aparente. Margens cirúrgicas livres. Paratireoide sem alterações. Estadiamento patológico – pT4.
Esvaziamento cervical é realizado e o laudo histopatológico evidencia infiltração neoplásica – Nível III D – presença de metástase para um dos quatro linfonodos isolados.
O exame genético do paciente identifica a mutação Cys634Arg no gene RET.
Por tratar-se de uma doença genética que pode desenvolver o carcinoma medular da tireoide, que é muito agressivo, realizou-se o rastreamento genético nos familiares através do exame de ácido desoxirribonucleico (DNA).
Abaixo seguem os diagnósticos gênicos – RET solicitados aos familiares:
– M.H. – (caso índice) Afetado, mutação Cys634Arg no gene RET
– E.H. – Normal (irmão)
– T.H. – Normal (irmã)
– M.H. – Normal (irmão)
– M.H. – Normal (irmão)
– B.H. – Normal (irmã)
– M.H. – Afetado, mutação Cys634Arg no gene RET (irmão)
– A.M. – Normal (irmã)
– A.H. – Normal (irmão)
– E.H. – Normal (filho)
– T.H. – Afetado, mutação Cys634Arg no gene RET (filha)
Os métodos utilizados nos diagnósticos: extração de DNA – amplificação gênica foram amplificados por Reação em Cadeia da Polimerase (PCR) os éxons 10, 11, 13, 14, 15 e 16 do gene RET, responsável pela Neoplasia Endócrina Múltipla, tipo 2. Identificação de Mutação: Sequenciamento gênico direto, capilar.
Discussão
Feocromocitomas são tumores raros localizados na medula suprarrenal, que desencadeiam crises hipertensivas devido à produção excessiva de catecolaminas. Normalmente esses tumores são benignos, e quando diagnosticados precocemente diminuem o quadro de mortalidade e/ou futuras complicações possíveis provocadas pela hipertensão arterial.(8)
As manifestações clínicas do feocromocitoma são semelhantes às encontradas em casos já estudados.(2,8) Em pacientes diagnosticados, os principais sintomas apresentados são: hipertensão, palpitações, cefaleia, ansiedade, sudorese e palidez.(9,10) Essas manifestações comuns com outras patologias podem retardar o diagnóstico, prejudicando o prognóstico do paciente. Esse tipo de neoplasia pode levar a crises hipertensivas severas, potencialmente letais, por esse motivo é importante investigar, tratar e ressecar esses tumores. A detecção eficaz desses casos leva ao diagnóstico precoce de possíveis tumores malignos.(8)
No presente relato de caso, o paciente iniciou os sintomas aos 32 anos de idade, a manifestação mais frequente foi a hipertensão arterial, seguido de cefaleia intensa, o que levou a equipe multidisciplinar que o recebeu no setor de emergência a suspeitar de meningite, e foi solicitada uma série de exames para investigação. O diagnóstico de feocromocitoma pode ser feito inicialmente por meio da clínica, exames de laboratório e de imagens.(11)
Vários relatos de casos foram estudados para identificar qual o melhor método deve ser utilizado no diagnóstico do paciente com o objetivo de evitar as manifestações severas dessa patologia.
Segundo Satendra e Tamayo,(10,11) os exames de imagens, tomografia axial computadorizada (TAC) e ressonância magnética (RM) apresentaram sensibilidade e especificidade superiores a 90%, tornando-se os exames de primeira escolha por permitir a localização adequada no tórax, abdômen e pelve, mas sua especificidade é limitada. A cintilografia com metaiodobenzilguanidina (MIBG) possui uma sensibilidade de 77%-91% e uma especificidade de 96% a 100% tornando-se o teste de escolha para localização de tumores.
Devido à variabilidade dos sintomas apresentados no diagnóstico do feocromocitoma, observa-se a importância dos exames de imagens e o aumento de achados incidentais do tumor, porém o diagnóstico por incidentalomas depende da idade do paciente e do tipo de exame de imagem solicitado.(2,3)
Os níveis plasmáticos e urinários elevados de catecolaminas e dos metabólitos, as metanefrinas, são testes de primeira linha para o estabelecimento do diagnóstico confirmatório em doentes com suspeita de feocromocitoma, pois a sensibilidade desses testes aproxima-se dos 100%.(12) Porém, não são precisos por diversos fatores como uso de alguns fármacos, estresse, tabagismo, consumo de café e de chocolate que podem aumentar a liberação de catecolaminas podendo, portanto, apresentar resultados falso-positivos.(13)
Os testes realizados na urina 24 horas de ácido vanilmandélico (VMA), as metanefrinas (totais e fracionadas) e as catecolaminas são testes iniciais e usados com frequência, porém as metanefrinas fracionadas e as catecolaminas apresentam maior sensibilidade. Já as dosagens de catecolaminas e metanefrinas plasmáticas são mais indicadas, entretanto as medições das metanefrinas plasmáticas são as mais sensíveis e menos propensas a apresentar resultados falso-positivos em consequência de estresse, incluindo a punção venosa.(12)
O feocromocitoma, em 56% dos casos, apresenta mutações no DNA, sendo indicados estudos genéticos em todos os pacientes, porém, em casos de histórico familiar positivo, o exame é imprescindível.(13)
A neoplasia endócrina múltipla (NEM 2A) é uma síndrome genética caracterizada pela presença do feocromocitoma, carcinoma medular da tireoide (CMT) e neoplasia das paratireoides, e 10% a 15% dos casos ocorrem sob a forma de carcinoma medular da tireoide familiar (CMTF).(14)
Sendo assim, o rastreamento genético deve ser realizado em todos os familiares por meio do diagnóstico molecular, que apresenta maior superioridade ao diagnóstico clínico e/ou bioquímico na identificação de indivíduos assintomáticos, em risco de desenvolvimento da neoplasia, pois 4% a 10% dos CMT apresentam mutações germinativas do RET.(15)
Dessa forma, o teste de rastreamento molecular do proto-oncogene RET torna-se primordial na avaliação diagnóstica e no planejamento terapêutico dos pacientes.(15)
Conclusões
Os feocromocitomas são tumores compostos de células cromafins que sintetizam e liberam catecolaminas. Os portadores dessa neoplasia apresentam sintomas clássicos como hipertensão arterial, cefaleia, sudorese e taquicardia, decorrentes da excreção de catecolaminas. Esses sintomas são comuns a diversas comorbidades, o que dificulta o diagnóstico precoce. Destaca-se a importância dos exames bioquímicos e de imagem na confirmação e localização do tumor. A identificação da mutação por meio de exames genéticos possibilita o rastreamento genético familiar, o monitoramento e a retirada de tumores antes do seu desenvolvimento.
A excisão do tumor por meio cirúrgico implica uma recuperação rápida e menor prevalência de morbimortalidades.
Abstract
Pheochromocytomas are tumors of chromaffin cells in the adrenal medulla that produce, store, metabolize, and secrete catecholamines. Due to increased secretion the patient may present with various symptoms, but elevation of blood pressure is the most common manifestation, and can cause serious complications if not recognized and treated in time. The present report mentions a case of a young male patient who started the picture with severe hypertension, tachycardia and headache, diagnosed with Pheochromocytoma during the investigation of paroxysmal arterial hypertension. It should be noted that early diagnosis, followed by surgical treatment of tumor removal, makes it possible to cure the disease and remission of symptoms.
Keywords
Pheochromocytoma; Arterial Pressure; Catecholamines
REFERÊNCIAS
- Igaki J, Nishi A, Sato T, Hasegawa T. A pediatric case of pheochromocytoma without apparent hypertension associated with von Hippel-Lindau disease. Clin Pediatr Endocrinol [Internet]. 2018;27(2):87-93. Disponível em: https://www. jstage.jst.go.jp/article/cpe/27/2/27_2017-0036/_article
- Teixeira BL, Bernardo MO. Achado incidental de feocromocitoma após exame de imagem. Rev da Fac Ciências Médicas Sorocaba [Internet]. 2017 Nov 13;19(3):154. Disponível em: https://revistas.pucsp.br/index.php/RFCMS/article/view/28669
- Marques AP, Paiva I, Sapinho I, Belo S, Couto J, Azevedo T, et al. Feocromocitoma: estudo retrospectivo multicêntrico. Rev Port Endocrinol Diabetes e Metab [Internet]. 2016;11(2):156-62. Disponível em: http://linkinghub.elsevier.com/retrieve/pii/S 1646343916300062
- Herdy GVH, Olivaes MC, Lopes VGS, Pontes CAG, Ormond Filho JB, Fonseca EC. Feocromocitoma em Criança Pheochromocytoma in Childhood. Soc Bras Cardiol. 2005;84:2004-6.
- Ortiz-Vázquez IC, Ramos-García MA, Juárez GM, Clavellina-Rosas JM, Moreno-Vázquez A, Calderón-abbo M. Infarto agudo del miocardio relacionado con feocromocitoma. Rev Medica del Seguro Soc. 2012;50(5):559-63.
- Ferreira MA, Vilaverde J. A genética dos feocromocitomas e paragangliomas. Rev Port Endocrinol Diabetes e Metab [Internet]. 2014;9(1):29-35. Disponível em: http://linkinghub.elsevier.com/retrieve/pii/S164634391400011X
- Megías MC, Puyol DR, Rodríguez LF, Martinez GLS, Miguel PM. Feocromocitoma-paraganglioma: del diagnóstico bioquímico al genético. Nefrología [Internet]. 2016 Sep;36(5):481-8. Disponível em: http://dx.doi.org/10.1016/j.nefro.2016.03.010
- Rubio-Marín AC, Orjuela AD, Rascovsky M, Rosselli D. Hipertensión secundaria a paraganglioma- presentación de un caso y revisión de la literatura. 2016;29(2):206-17.
- Sanabria C, Vendrell M. Severe cardiomyopathy secondary to pheochromocytoma: Usefulness of magnesium sulfate. Case report. Rev Colomb Anestesiol. 2016;44:58-62. Disponível em: http://www.scielo.org.co/scielo.php?script=sci_arttext&pid=S0120-33472016000100013&lng=en.
- Satendra M, Jesus C, Armando L, Sá ALB, Rosário L, Rocha J, et al. Reversible catecholamine-induced cardiomyopathy due to pheochromocytoma: Case report. Rev Port Cardiol. 2014;33(3). DOI: 10.1016/j.repce.2013.09.014
- Uribea JD, González MR, Tamayo LJ. Manifestaciones inusuales del feocromocitoma. Reporte de caso y revisión de la literatura. Rev. Colomb. Cardiol. [online]. 2016, vol.23, n.2, pp.151.e1-151.e5. http://dx.doi.org/10.1016/j.rccar.2015.06.013.
- Gomes C, Laranjo G, Santos E, Faria C. Quando hipertensão arterial persistente no adolescente tem uma origem endócrina rara: relato de dois casos e revisão da literatura. SciMed (Porto Alegre). 2016;26(4):4-11.
- Chen Y, Xiao H, Zhou X, Huang X, Li Y, Xiao H, et al. Accuracy of plasma free metanephrines in the diagnosis of pheochromocytoma and paraganglioma: a systematic review and meta-analysis. Endocr Pract [Internet]. 2017 Oct;23 (10):1169-77. Disponível em: http://journals.aace.com/doi/10.4158/EP171877.OR
- Rodrigues P, Castedo JL. Síndrome MEN Tipo 2. Arq Med. 2012;26(6):239-44. Disponível em: http://www.scielo.mec. pt/scielo.php?script=sci_arttext&pid=S0871-3413201 2000600002&lng=pt.
- Maia AL, Siqueira DR, Kulcsar MAV, Tincani AJ, Mazeto GMFS, Maciel LMZ. Diagnosis, treatment, and follow-up of medullary thyroid carcinoma: recommendations by the Thyroid Department of the Brazilian Society of Endocrinology and Metabolism. Arq Bras Endocrinol Metabol. 2014 Oct;58(7):667-700. [Article in En, Portuguese]. doi: 10.1590/0004-2730000003427.
Correspondência
Jaqueline Antunes de Ramos
Av. Getúlio Vargas, 268 – Centro
89251-970 – Jaraguá do Sul-SC, Brasil