Novas perspectivas no diagnóstico da policitemia vera: uma revisão sistemática da literatura
New perspectives in diagnosis of polycythemia vera: a systematic review
Elizane Pinheiro Monteiro1, Isaura Abreu Maciel1, Eliane Pereira de Souza1, Symara Rodrigues Antunes1, Danielle Cristinne Azevedo Feio1
1 Centro Universitário Metropolitano da Amazônia – UNIFAMAZ, Curso de Biomedicina. Belém, PA, Brasil.
Recebido em 11/10/2022
Aprovado em 07/11/2022
DOI: 10.21877/2448-3877.202200077
INTRODUÇÃO
A policitemia vera é uma neoplasia hematológica não leucêmica, também classificada como uma neoplasia mieloproliferativa (NMP). Caracteriza-se por ser uma doença clonal da célula estaminal hematopoiética, associada a mutações no gene JAK2V617F em quase todos os casos. Desta forma, resulta na desregulação da transdução de sinal, como consequência de mutação somática adquirida, geralmente em genes que codificam a proteína tirosina quinase.(1) Além disso, a célula hematopoiética multipotente irá multiplicar-se sem controle, gerando grande quantidade de células filhas com a mesma mutação, causando assim a neoplasia maligna.(2)
A mutação no gene JAK2 se dá no interior do domínio JH2 (Janus homology 2), do éxon 14, no nucleotídeo 1849, o que resulta na substituição de valina por fenilalanina. É uma mutação com ganho de função em que se perde a ação autoinibitória de JH2 e na expressão de uma tirosina quinase JAK2 ativada. A mutação JAK2V617F ocorre em mais de 95% dos casos de PV; os 5% restantes apresentam mutações no éxon 12 do gene JAK2, o que acontece mais raramente.(3)
Ao longo do curso natural da doença, a PV apresenta alterações e várias complicações, com redução da expectativa de vida do indivíduo acometido. Ainda nos dias atuais, a estratégia de tratamento é limitada, baseada sobretudo no diagnóstico diferencial individualizado. No entanto, o sinal clínico mais comum entre os portadores de PV (aumento de eritrócitos), por si só, é insuficiente para o diagnóstico definitivo, além de a variabilidade fenotípica ser um fator que obriga a inclusão de mais critérios para se chegar a uma conclusão diagnóstica definitiva.(4)
Desta forma, o objetivo deste trabalho é realizar um estudo exploratório para abordar novas perspectivas no diagnóstico diferencial, assim como esclarecer quais os métodos utilizados para tal fim, além de informar as mutações genéticas que possam auxiliar e contribuir para o painel de exames da PV e diferenciá-la de outros distúrbios mieloproliferativos.
METODOLOGIA
Este trabalho, que se caracteriza como uma revisão sistemática da literatura, realizou um estudo exploratório com ênfase no diagnóstico da policitemia vera e seguiu o protocolo Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA).(5) A seleção dos artigos científicos foi feita por meio de buscas nas bases de dados eletrônicos: National Library of Medicine (PubMed), BVS Brasil (Biblioteca Virtual em Saúde – Brasil) e SciELO (Scientific Electronic Library Online). Foram utilizados para a consulta descritores em inglês, sendo estes: ‘‘polycythemia vera diagnosis” e “polycythemia vera mutations”, e os mesmos descritores em português “policitemia vera” e “policitemia vera mutação”, em todas as bases de dados utilizadas. Em relação aos filtros nas plataformas de busca foram adotados os de idioma (inglês e português), espécie (humano) e ano (2014 a 2021) de publicação.
A seleção dos artigos incluídos na revisão foi realizada por três autores de forma independente, focando estudos potencialmente relevantes; um quarto revisor era requerido quando não havia consenso. Os estudos passaram por triagem com base na leitura do título, resumo e, posteriormente, a análise na íntegra de cada artigo, aplicando sempre os critérios de exclusão e inclusão descritos a seguir.
Os seguintes critérios de inclusão foram adotados: estudos somente com humanos, artigos de pesquisa, artigos disponíveis na integra, artigos que abordassem os diagnósticos laboratoriais associados ou não às mutações, e artigos publicados no período de 8 anos (intervalo entre 2014 a 2021). Já os critérios de exclusão foram: estudos em animais, artigos duplicados, carta ao editor, entrevistas, artigos de revisão, ensaios clínicos, relato de caso e artigos que não abordassem a temática proposta neste estudo.
RESULTADOS
A pesquisa bibliográfica realizada nas três bases de dados, com os descritores anteriormente estabelecidos, contabilizou 2.183 artigos, dos quais 1.362 foram encontrados na base de dados eletrônico National Library of Medicine (PubMed), 821 na base de dados BVS Brasil (Biblioteca Virtual em Saúde Brasil) e 0 na base de dados SciELO (Scientific Electronic Library Online). Após as duas primeiras etapas foram excluídos 1.848 artigos por não abordarem a temática da pesquisa e selecionados 320 artigos para a avaliação de elegibilidade; destes, 25 artigos foram incluídos em síntese quantitativa para compor a revisão (Figura 1).
Em relação ao perfil das publicações selecionadas para a revisão de acordo com o ano e o quantitativo de trabalhos, verificou-se que dos 25 estudos incluídos que foram selecionados entre os anos de 2014 a 2021, cinco (20%) eram de 2014, quatro (16%) de 2015, seis (24%) de 2016, um (4%) de 2017, três (12%) de 2018, um (4%) de 2019, três (12%) de 2020 e dois (8%) de 2021.
A Tabela 1 sintetiza as informações retiradas dos 25 artigos qualificados para integrar os resultados da pesquisa, bem como seus respectivos autores, ano, revista, tipo de estudo e principais resultados de cada um dos trabalhos.
A partir da análise dos novos diagnósticos propostos, foram selecionadas as principais proposições de alterações dos parâmetros de diagnóstico laboratorial, que representam as perspectivas mais promissoras de mudanças dos critérios para o diagnóstico da PV (Tabela 2).
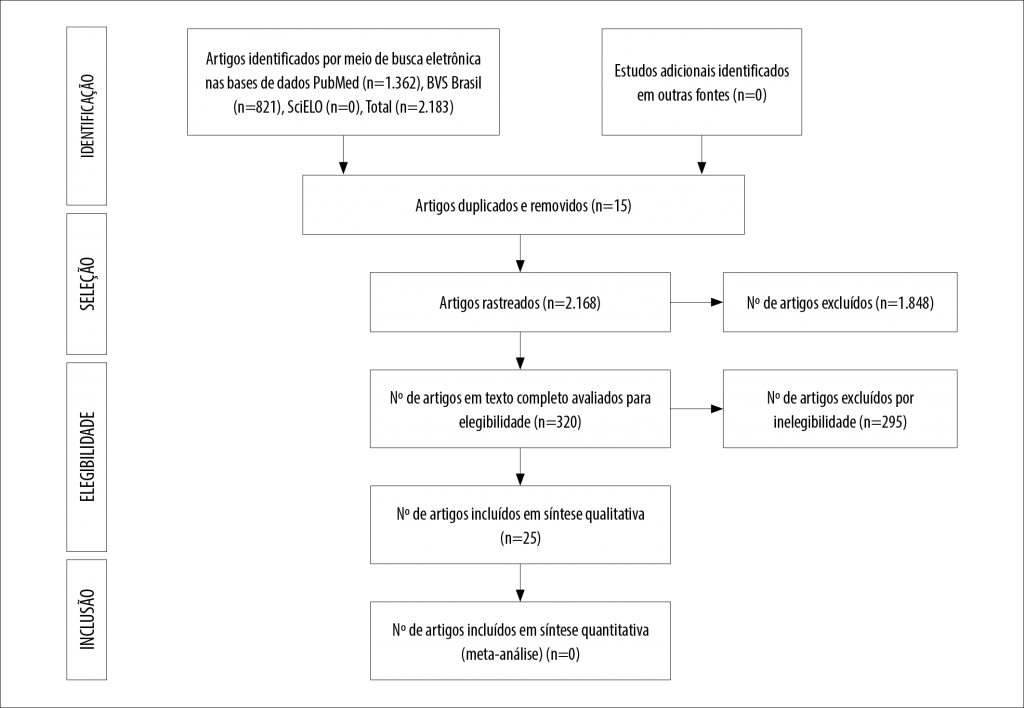
Figura 1
Fluxograma com os critérios de busca eletrônica em três bases de dados.
Tabela 1
Apresentação do material qualificado para integrar os resultados da pesquisa.
| Autor, ano | Revista | Tipo de estudo | Principais resultados |
| Davila-Gonzalez et al., 2021(6) | Clinical Lymphoma Myeloma and Leukemia | Retrospectivo | O diagnóstico PV usando JAK2V617 é mais eficaz que o teste EPO. Um nível de EPO < 2 mIU/mL foi > 99% específico para prever PV, mas foi apenas 12% sensível. |
| Zakaria et al., 2021(7) | The International Journal of Laboratory Hematology | Quantitativo | Presença de mutações para CALR em paciente PV com p.K368del (c .1102_1104delAAG) e mutações do inframe de inserção (c.1135insA). |
| Nersesjan et al., 2020(8) | European Journal of Hematology | Retrospectivo | Uma boa correlação da contagem total de eritrócitos versus HTC foi encontrada para indivíduos com PV quando indivíduos com microcitose foram excluídos (R2 = 0,87). Especificidade de 98% e sensibilidade de 37% para VHS < 2mm no diagnóstico de PV. |
| Lupak et al., 2020(9) | Blood Cells, Molecules, and Diseases | Retrospectivo | 32% dos pacientes com PV apresentaram níveis de EPO dentro da normalidade, sendo que o nível de EPO correlacionou-se positivamente com obesidade e tabagismo. |
| Cacemiro et al., 2020(10) | Scientific Reports | Quantitativo | Pacientes com PV em comparação com o grupo controle e com pacientes com policitemia secundária apresentaram níveis aumentados de GM-CSF, IFN-α2, IFN-γ, IL-12p70, IL- 17A, IL-5, IP-10, MIP-1α, MIP-1 e TNF-α. |
| Trung et al., 2019(11) | BMC Medical Genetics | Quantitativo | PCR-ALDA (Polymerase Chain Reaction based amplicon length differentiation assay) pode ser utilizado como uma ferramenta fácil de usar, rápida, de baixo custo e sensível na detecção de mutações CALR em pacientes com NMP, incluindo a PV. |
| Senín et al., 2018(2) | Annals of Hematology | Quantitativo | Pacientes com PV que progrediram para LMA apresentaram elevada frequência de mutações adicionais: DNMT3A, TET2, ASXL1 por NGS. |
| Jakovic et al., 2018(12) | Annals of Hematology | Retrospectivo | 213 pacientes com PV mostraram valores de HB: 18,4 – 19,7g/dL e 240 pacientes mostraram níveis de HTC aumentado: 55,3 – 60,7%. |
| Spivak, 2018 (3) | Leukemia | Descritivo | Para pacientes de PV a conduta mais adequada é a medição da carga alélica de JAK2V617F por PCR. |
| Tefferi e Barbui, 2017 (13)
|
American Journal of Hematology | Qualitativo | Redução do limiar de HB e HTC para 16,5 g/dL e 49% para homens; e 16g/dL e 48% para mulheres, respectivamente. |
| Kander et al., 2016(14) | Journal of the National Comprehensive Cancer Network | Qualitativo | Os testes de diagnóstico mais utilizados: células vermelhas (57%), níveis de eritropoietina (83,1%) e teste para mutações no gene JAK2 (90,1%). |
| Wang et al., 2016(15) | Journal Plos One | Quantitativo | Pacientes com PV não tratados tinham níveis mais elevados de RFCI-1 do que os pacientes com policitemia secundária e pacientes com PV tratados. |
| Alghasham et al., 2016(18) | International Journal of Laboratory Hematology | Quantitativo | A análise de mutações de 271 pacientes indicou 262 positivos para a mutação JAK2V617F e 09 para mutação não positiva realizada através do sequenciamento de Sanger associado com o PCR. |
| Reiter e Harrison, 2016(16) | Current Hematologic Malignancy Reports | Descritivo | O ruxolitinib pode ser um tratamento eficaz para a leucocitose persistente e trombocitose, com leucócitos ≤15 x109/L, contagem de plaquetas ≤600 x109/L, normalização em HTC, leucócitos e plaquetas. |
| Almedal et al., 2016(17) | Tidsskr Nor Legeforen | Quantitativo | O método de PCR indicou que a mutação V617F positiva foi a mais frequente, e demonstrou níveis de plaquetas >450.000. |
| Leszczynska et al., 2016(19) | Acta Haematologica | Quantitativo | Os métodos utilizados para a identificação da mutação foram: amplificação refratária e PCR, com identificação positiva em 91% dos casos. |
| Yigit et al., 2015(20) | Human Pathology | Exploratório | Os casos de PV tiveram maior frequência de eritroblastos nucleares positivos. Expressão elevada de FEN2 foi vista em TE e PV 50% ± 13,3% e 41,5% ± 9,4%, respectivamente. |
| Tefferi e Barbui, 2015 (21) | American Journal of Hematology | Descritiva | Alteração nos valores de HB e HTC de HB> 18,5g/dL em homens e > 16,5g/dL em mulheres para HB> 16,5g/dL em homens e > 16,0g/dL em mulheres; HTC > 49% em homens e > 48% em mulheres. |
| Barbui et al., 2015(1) | American Journal of Hematology | Retrospectivo | Os valores de HB e HTC mudaram de 18,5 para 18,1 e de 55,6 para 54,7, respectivamente, após a descoberta da mutação JAK2V617F. |
| Angona et al., 2015(22) | European Journal of Haematology | Quantitativo | Mutações adicionais; TET2, DNMT3A através do NGS. |
| Barbui et al., 2014 (23) | American Journal of Hematology | Descritivo | Os níveis de HB (<16,0 g/dL) e HTC (48%) foram mais baixos em CALR contra pacientes JAK2 mutado; o HB e HTC de PV foram mais elevados: HB >19,0g/dL e HTC 52%. |
| Roda et al., 2014(24) | Annals of Hematology | Retrospectivo | 98% dos pacientes testados para PV apresentaram a mutação JAK2V617F. |
| Barbui et al., 2014(25) | American Journal of Hematology | Descritivo | Pacientes apresentaram contagem de plaquetas >450.000 e aumento da fibrose reticular na medula óssea (hipercelularidade). |
| Ouyang et al., 2014(26) | Clinical Cytometry | Retrospectivo | Em comparação com as SM, as NMP Ph-negativos exibem alterações imunofenotípicas menos frequentes, tais como alterações na expressão de blastos CD341 na MO. |
| Ancochea et al., 2014 (27) | British Journal of Haematology | Retrospectivo | O nível sérico de EPO foi medido em 106 pacientes, e correspondeu a valores <10 mUI/mL do limite inferior de normalidade para cada teste JAK2V617F positivo. |
Policitemia Vera (PV); Organização Mundial de Saúde (OMS); Hemoglobina (HB); Hematócrito (HTC); Velocidade de hemossedimentação (VHS); Neoplasias Mieloproliferativas (NMPs); Clínica Europeia Molecular e Patológica (ECMP); Sequenciamento de nova geração (NGS); Medula Óssea (MO); Leucemia Mieloide Aguda (LMA); Eritropoietina (EPO); Síndromes Mielodisplásicas (SM); Fator Eritroide Nuclear 2 (FEN2); Receptor do Fator de Crescimento Semelhante à Insulina 1 (RFCI-1); Calreticulina (CALR).
Tabela 2
Proposições de alterações nos parâmetros laboratoriais para o diagnóstico da policitemia vera.
| Teste Laboratorial | Técnica | Parâmetro Atual | Novo Parâmetro | Parâmetro/Técnica substituído |
| Pesquisa de anormalidades em células mielomonocíticas | Citometria de Fluxo | ——- | Alterações imunofenotípicas e na maturação celular | ——- |
| Quantificação dos níveis de RFCI-1 | Citometria de Fluxo | ——- | 227,8 – 461,1 | Biópsia da MO |
| Pesquisa de FEN2 | Imuno-histoquímica | ——- | Positividade para FEN2 e nível de coloração nuclear >30% | ——- |
| Análise da estrutura dos genes | Sequenciamento de Sanger | Detecção JAK2 | TET2, DNMT3A | NGS |
| Detecção de variações do genoma, análise de expressões de genes | NGS | Detecção JAK2 | JAK2V617F positiva e negativa, DNMT3A, TET2, ASXL1 | ——- |
NI: não informado; RFCI-1: Receptor do fator de crescimento semelhante à insulina 1; MO: Medula Óssea; FEN2: Fator eritróide nuclear 2; CEIA: Método de enzima imunoquimioluminescente melhorada; NGS: Sequenciamento de nova geração.
DISCUSSÃO
A policitemia vera é caracterizada pelo aumento da massa de glóbulos vermelhos, com o aspecto morfológico normal das células. Os níveis de eritropoietina (EPO) possuem valores normais ou baixos, porém os valores do hematócrito (HTC) são anormais, com hematopoiese extramedular, hiperviscosidade sanguínea e grandes riscos de complicações como trombose venosa ou arterial e hemorragia, além de risco de evolução para mielofibrose ou transformação em leucemia mieloide aguda.(17,19)
Segundo estudo de Leszczynska et al.(19) e Spivak,(3) o gene JAK2 está localizado no cromossomo 9p24 e codifica uma tirosina quinase no receptor que está distribuído no citoplasma da célula, e esta é a via de transdução de sinal que regula processos celulares como proliferação, diferenciação e apoptose. A mutação mais frequentemente adquirida é encontrada no gene JAK2 do éxon 14 no nucleotídeo 1849, o que resulta na substituição de valina por fenilalanina, localizada no domínio pseudoquinase (JH2), e isso leva à ativação da via de sinalização, induz o crescimento independente de citocina do progenitor hematopoiético e o aumento da produção das células da linhagem mieloide. Esta mutação é encontrada na maioria dos pacientes com policitemia vera V617F positivo com a utilização das técnicas de PCR.
Vários autores discutem o diagnóstico de PV por meio da detecção da mutação JAK2V617F através de sequenciamento de Sanger. Dentre eles, destacam-se os resultados encontrados por Alghasham et al.,(18) os quais informam que a mutação JAK2V617F positiva foi mais prevalente (96,7%). Já Almedal et al.,(17) ao utilizarem a mesma técnica, demonstram uma prevalência proporcionalmente menor (66,7%) na mutação JAK2V617F positiva e que as mutações JAK2V617F negativas (prevalência de 33,3%) foram identificadas nos éxons 12 e 13. Soma-se a isso o fato de que a mutação JAK2 provou ser um importante marcador no diagnóstico de neoplasias mieloproliferativas.
De acordo com Senín et al.,(2) existe uma grande instabilidade genética clonal em pacientes com PV JAK2V617F positiva, uma vez que essa mutação é altamente variável. Assim sendo, os pacientes que desenvolvem leucemia aguda são caracterizados por apresentarem mutações adicionais, como DNMT3A, TET2 e ASXL1, dados esses obtidos através da técnica de sequenciamento de nova geração (NGS). Este fato está em concordância com o estudo de Angona et al.,(22) no qual os pacientes que apresentaram dominância clonal ao nível celular e que evoluíram para outras neoplasia, foram observadas mutações adicionais: ASXL1 no éxon 12 utilizando NGS, e mutações TET2, DNMT3A por sequenciamento de Sanger.
A presença dessas mutações adicionais identificadas pelo sequenciamento de Sanger e NGS tem implicações importantes e imediatas no diagnóstico da PV, e consequentemente na possibilidade de redução da progressão de PV para a LMA. Tais achados corroboram a evolução e a importância das aplicações do diagnóstico molecular no processo de diferenciação da PV.
A partir de 2005 a mutação V617F começou a ser usada como marcador para o diagnóstico de PV, além do que, os valores de HB e HTC mudaram de 18,5 para 18,1 e de 55,6 para 54,7, respectivamente. Desde então, houve significativa redução nas estatísticas de trombose arterial e venosa (de 1.00 para 0.80) e aumento da taxa de sobrevida resultantes de terapia e de acompanhamento adequados.(1)
Após essa constatação, diversos autores buscaram modificar os parâmetros ou acrescentar outros exames ao diagnóstico da PV. Roda et al.(24) afirmam que o diagnóstico da PV deve se basear em uma avaliação que abranja as características clínicas e laboratoriais, incluindo estado e quantificação da mutação da JAK2, como também o nível de eritropoietina no soro. Assim sendo, a presença da mutação JAK2V617F e um nível anormal de EPO no soro confirmam o diagnóstico de PV. Já na presença de níveis baixos de EPO no soro e ausência de JAK2V617F, o autor sugere fazer uma análise adicional para mutações JAK2 éxon 12. Deste modo, a biópsia da MO torna-se um teste não essencial para o diagnóstico.
Entretanto, Lupak et al.(9) observaram que considerar apenas o nível de EPO não promove um marcador diagnóstico confiável devido à variação fisiológica em associação com obesidade e tabagismo. Tal achado correlaciona-se com o visto por Davila-Gonzalez et al.(6) que afirmam que os valores séricos de EPO podem não ser úteis como única ferramenta de triagem para diagnosticar PV ou diferenciar da eritrocitose idiopática, pois sobretudo no cenário clínico apropriado, com estudos citogenéticos e moleculares, como o status da mutação JAK2V617F, esses prevalecem como as ferramentas mais úteis para a detecção de PV.
Kander et al.(14) afirmam que a maioria dos profissionais hematologistas utiliza os critérios da OMS para diagnosticar seus pacientes com suspeita de PV. A estratégia de diagnóstico mais comum entre eles é o teste para a mutação JAK2V617F seguido da avaliação do nível de EPO no soro aspirado da MO e quantificação da massa de glóbulos vermelhos.
A OMS estabelece como critério menor para o diagnóstico de PV os níveis baixos (<10mUI/mL) de EPO no soro do paciente. Ancochea et al.(27) avaliaram a precisão deste critério, quando associado à quantificação da carga mutacional e à leitura qualitativa do HTC, a partir do método de enzima imunoquimioluminescente melhorada (CEIA). Os resultados mostraram que a precisão dos níveis de EPO medidos de forma isolada é alta. Entretanto, em relação aos níveis de EPO, quando avaliadas juntamente com a quantificação da carga alélica mutacional do JAK2V617F positivo, mostrou uma sensibilidade e especificidade de 83% a 85%, respectivamente. Por fim, a junção de resultados da carga alélica mutacional e da leitura qualitativa do HTC (>48%), a sensibilidade e especificidade chegou em 79% e 97%, respectivamente. Com isso, o autor sugere adicionar aos critérios menores a quantificação da carga alélica mutacional, pois este parâmetro em junção com outros menores tem alta precisão.
De acordo com Ouyang et al.,(26) as NMP apresentam alterações imunofenotípicas frequentes, como anormalidades em células CD341 ou em outras células mielomonocíticas, mais acentuadas nos casos com características histopatológicas adversas. Estas observações são feitas com mais precisão através do método de imunofenotipagem por citometria de fluxo (ICF), que tem sido um exame de alta relevância na pesquisa de malignidades hematopoiéticas, por mostrar com especificidade células com fenótipo anormal.
Com base no exposto, Wang et al.(15) propuseram quantificar os níveis do receptor do fator de crescimento semelhante à insulina 1 (RFCI-1), através de citometria de fluxo. Os resultados mostraram que nenhum dos pacientes com policitemia secundária ou controles normais tinha níveis aumentados de RFCI-1, ao passo que 87% dos pacientes com PV apresentaram níveis significativamente elevados no sangue periférico, o que evidencia que o aumento dos níveis desse hormônio é um sinal característico de pacientes com PV. Desta forma, a quantificação de RFCI-1 pode substituir a utilização da formação de colônias eritroides endógenas, utilizadas como critério menor pela OMS, visto que para realizar tal processo é necessária a aspiração da MO, em contraste com a coleta de sangue periférico usada para medir os níveis de RFCI-1. Desta forma, a utilização deste meio reduziria o custo e o tempo de análise necessários para se chegar ao diagnóstico.
Yigit et al.(20) avaliaram através de imuno-histoquímica as diferenças na expressão de fator eritroide nuclear 2 (FEN2), receptor do fator de crescimento do nervo, p53, CD34, CD68, CD3, CD20 e CD138 nas três principais síndromes mieloproliferativas: trombocitemia essencial (TE), mielofibrose primária (MFP) e PV. Foi observado que pacientes com TE e PV têm maior expressão nos níveis de FEN2, frequências mais elevadas de eritroblastos nucleares positivos e que a contagem de células T pode ser útil para discriminar a TE de PV. Além de que, se o nível de coloração do núcleo de eritoblastos positivos para FEN2 (>30%) for utilizado como critério de corte, pode-se excluir a possibilidade de MFP, o que reduz e facilita a conclusão do diagnóstico diferencial. Pode fornecer também uma nova maneira de avaliar biópsias de pacientes portadores de algum distúrbio mieloproliferativo, visto que este resultado favorece o diagnóstico de TE e de PV. A intenção é chegar mais rapidamente ao diagnóstico e, se possível, substituir alguns dos critérios menores utilizados pela OMS para o diagnóstico de PV.
Em relação ao diagnóstico diferencial da PV e da policitemia secundária (PS), é interessante a utilização de biomarcadores de oncoinflamação, uma vez que os pacientes com PV apresentam níveis plasmáticos aumentados das citocinas IL-17A, IFN-γ, IL-12p70 e TNF-α, em comparação a indivíduos saudáveis e com PS. Valendo ressaltar que os níveis mais elevados de IL-17A só foram detectados em pacientes com PV quando utilizada a plataforma multiplex, fato que não se repetiu ao usar a metodologia ELISA.(10)
Para Barbui et al.,(23,25) os diagnósticos da PV e da policitemia vera mascarada (PVM) necessitam dos critérios da OMS e do Comitê Britânico de Padrões em Hematologia (CBPH) para a sua caracterização, usando a comparação dos valores de HTC e de HB com a expressão de JAK2V617F, JAK2 éxon 12, e mutações na Calreticulina (CALR). O que também é corroborado pelos achados de Mutações de calreticulina (CALR) visto através de diferentes técnicas de diagnóstico por Zakaria et al.(7) e Trung et al.(11)
O estudo de Tefferi e Barbui(13) relata que alterações nos níveis limiares de HB e HTC da OMS de 2016 devem ser feitas para incluir pacientes com PVM nos critérios de diagnóstico, com modificação nos parâmetros de HB > 18,5g/dL em homens e > 16,5g/dL em mulheres para HB > 16,5g/dL em homens e > 16,0g/dL em mulheres; HTC > 49% em homens e > 48% em mulheres. Segundo eles, esses níveis são valores de corte ideais para distinguir pacientes portadores de PVM. O diagnóstico também pode ser associado aos resultados da biópsia da MO que sejam compatíveis com a PV.
Além do exposto, Nersesjan et al.(8) propõem uma combinação da contagem total de eritrócitos e a VHS (velocidade de hemossedimentação) como uma nova ferramenta para substituir a concentração de HB e o HTC no diagnóstico de PV, visto que esta estratégia pode refletir mais precisamente o estado de hipercoagulabilidade do paciente com PV. Nesse sentido, vale ressaltar que foi encontrada uma boa correlação de contagem total de eritrócitos versus HTC para indivíduos com PV quando pacientes com microcitose foram excluídos (R2=0,87), além de especificidade de 98% e sensibilidade de 37% para VHS < 2mm no diagnóstico de PV.
Para Tefferi e Barbui(13) e para Jakovic et al.,(12) os pacientes com PV não só apresentam manifestações clássicas da doença, com confirmação pela OMS, como também podem apresentar baixos níveis de EPO e níveis de HB ou HTC limítrofes. Relatam ao mesmo tempo que exibem resultados semelhantes, como contagem alta de plaquetas e o aumento da fibrose reticular na MO. Estas descobertas podem ter um impacto significativo sobre a classificação e terapia de tratamento.
Diante disso, Reiter e Harrison(17) expõem a importância de um diagnóstico rápido e preciso, para uma resposta adequada às terapias atuais. Ruxolitinib, o primeiro inibidor de JAK1 e JAK2, é o medicamento mais indicado para o tratamento de pacientes com PV, agora aprovado para pacientes com resistência ou intolerância à hidroxiureia. Tefferi e Barbui(21) ressaltam que os inibidores de JAK podem ser mais eficazes no alívio da carga mutacional e na melhoria da qualidade de vida de portadores da doença, destacando ainda ser necessário que todos os pacientes com PV recorram à flebotomia para manter o HTC abaixo de 45%. Esses benefícios podem compensar o custo da terapia, pois o principal objetivo da terapia em PV é prevenir complicações trombo-hemorrágicas e aumentar a expectativa de vida do paciente.
CONCLUSÃO
As estratégias de diagnóstico para PV empregadas atualmente deixam margem no seu diagnóstico diferencial. Entretanto, novas abordagens vêm criando táticas diferenciadas no gerenciamento da doença e contribuindo para melhorar a sua classificação, como a detecção de anomalias do gene JAK2, em especial para a mutação JAK2V617F, assim como a quantificação de RFCI-1, a expressão de FEN2 elevados e a associação dos critérios menores estabelecidos pela OMS, com o nível de carga mutacional que o indivíduo apresenta. Tais inovações mostram-se promissoras para a redução do tempo de diagnóstico e podem contribuir positivamente para o painel de exames diferenciais de PV. Portanto, é necessário olhar cautelosamente para marcadores que possam colaborar com a atualização do diagnóstico e levar em consideração a possibilidade de alteração ou substituição de alguns critérios já estabelecidos. Pois quanto mais rápido e preciso for o diagnóstico diferencial, mais ágil será a tomada de decisão em relação ao tratamento mais adequado para cada paciente.
REFERÊNCIAS
- Barbui T, Vannucchi AM, Carobbio A, Thiele J, Rumi E, Gisslinger H, et al. Patterns of presentation and thrombosis outcome in patients with polycythemia vera strictly defined by WHO-criteria and stratified by calendar period of diagnosis. Am J Hematol. 2015 May;90(5):434-7. doi: 10.1002/ajh.23970.
- Senín A, Fernández-Rodríguez C, Bellosillo B, Camacho L, Longarón R, Angona A, et al. Non-driver mutations in patients with JAK2V617F-mutated polycythemia vera or essential thrombocythemia with long-term molecular follow-up. Ann. Hematol. 2018 Mar;97(3):443-451. doi: 10.1007/s00277-017-3193-5.
- Spivak JL. Polycythemia vera. Curr. Treat. Options Oncol. 2018 Mar 7;19(2):12. doi: 10.1007/s11864-018-0529-x.
- Raedler LA. Diagnosis and Management of Polycythemia Vera: Proceedings from a Multidisciplinary Roundtable. Am Health Drug Benefits. 2014;7(7 Suppl 3):S36–S47.
- Shamseer L, Moher D, Clarke M, Ghersi D, Liberati A, Petticrew M, et al. Preferred reporting items for systematic review and meta-analysis protocols (PRISMA-P) 2015: elaboration and explanation. BMJ. 2015 Jan 2;350:g7647. doi: 10.1136/bmj.g7647.
- Davila-Gonzalez D, Barrios-Ruiz A, Fountain E, Cheng L, Masarova L, Verstovsek S, Rojas-Hernandez CM. Diagnostic Performance of Erythropoietin Levels in Polycythemia Vera: Experience at a Comprehensive Cancer Center. Clin Lymphoma Myeloma Leuk. 2021 Apr;21(4):224-229. doi: 10.1016/j.clml.2020.11.002.
- Zakaria NA, Rosle NA, Siti Asmaa MJ, Aziee S, Haiyuni MY, Samat NA, Husin A, et al. Conformation sensitive gel electrophoresis for the detection of calreticulin mutations in BCR-ABL1-negative myeloproliferative neoplasms. Int J Lab Hematol. 2021 Dec;43(6):1451-1457. doi: 10.1111/ijlh.13628.
- Nersesjan V, Zervides KA, Sørensen AL, Kjaer L, Skov V, Hasselbalch HC. The red blood cell count and the erythrocyte sedimentation rate in the diagnosis of polycythaemia vera. Eur J Haematol. 2020 Jan;104(1):46-54. doi: 10.1111/ejh.13334.
- Lupak O, Han X, Xie P, Mahmood S, Mohammed H, Donthireddy V. The role of a low erythropoietin level for the polycythemia vera diagnosis. Blood Cells Mol Dis. 2020 Feb;80:102355. doi: 10.1016/j.bcmd.2019.102355.
- Cacemiro MDC, Cominal JG, Berzoti-Coelho MG, Tognon R, Nunes NS, Simões B, Pereira ÍS, et al. Differential cytokine network profile in polycythemia vera and secondary polycythemia. Sci Rep. 2020 Apr 27;10(1):7032. doi: 10.1038/s41598-020-63680-7.
- Trung NT, Quyen DT, Hoan NX, Giang DP, Trang TTH, Velavan TP, Bang MH, et al. Rapid, low cost and sensitive detection of Calreticulin mutations by a PCR based amplicon length differentiation assay for diagnosis of myeloproliferative neoplasms. BMC Med Genet. 2019 Jun 27;20(1):115. doi: 10.1186/s12881-019-0819-6.
- Jakovic L, Gotic M, Gisslinger H, Soldatovic I, Sefer D, Tirnanic M, et al. The WHO diagnostic criteria for polycythemia vera-role of red cell mass versus hemoglobin/hematocrit level and morphology. Ann. Hematol. 2018 Sep;97(9):1581-90. doi: 10.1007/s00277-018-3344-3.
- Tefferi A, Barbui T. Polycythemia vera and essential thrombocythemia: 2017 update on diagnosis, risk-stratification, and management. Am J Hematol. 2017;92(1):94-108. doi: 10.1002/ajh.24607.
- Kander EM, Moliterno AR, Rademaker A, Streiff MB, Spivak JL, Stein BL. Practice patterns in the diagnosis and treatment of polycythemia vera in the post-jak2 v617f discovery era. J Natl Compr Canc Netw. 2016 Oct;14(10):1238-1245.
- Wang JC, Shi G, Baptiste S, Yarotska M, Sindhu H, Wong C, et al. Quantification of IGF-1 receptor may be useful in diagnosing polycythemia vera suggestion to be added to be one of the minor criterion. Plos One. 2016 Nov;11(11):e0165299. doi: 10.1371/journal.pone.0165299.
- Reiter A, Harrison C. How we identify and manage patients with inadequately controlled polycythemia vera. Curr Hematol Malig Rep. 2016 Oct;11(5):356-67. doi: 10.1007/s11899-016-0311-8.
- Almedal H, Vorland M, Aarsand AK, Gronningsæter IS, Bruserud O, Reikvam H. Myeloproliferative neoplasms and JAK2 mutations. Tidsskr Nor Laegeforen. 2016 Dec 6;136(22):1889-94.
- Alghasham N, Alnouri Y, Abalkhail H, Khalil S. Detection of mutations in JAK2 exons 12–15 by sanger sequencing. Int. J. Lab. Hematol. 2016 Feb;38(1):34-41. doi.org/10.1111/ijlh.12425.
- Leszczynska A, Grzenkowicz-Wydra J, Chmielewska-Gorycka L, Bieniaszewska M, Hellmann A. Detection of JAK2 exon 12 mutations in JAK2 V617F-negative polycythemia vera patients by cloning technique. Acta Haematol. 2016;136(2):123-8. doi: 10.1159/000446798.
- Yigit N, Covey S, Barouk-Fox S, Turker T, Geyer JT, Orazi A. Nuclear factor-erythroid 2, nerve growth factor receptor, and CD34-microvessel density are differentially expressed in primary myelofibrosis, polycythemia vera, and essential thrombocythemia. Hum Pathol. 2015 Aug;46(8):1217-25. doi: 10.1016/j.humpath.2015.05.004.
- Tefferi A, Barbui T. Polycythemia vera and essential thrombocythemia: 2015 update on diagnosis, risk‐stratification and management. Am. J Hematol. 2015 Feb;90(2):162-73. doi: 10.1002/ajh.23895.
- Angona A, Alvarez-Larran A, Bellosillo B, Martínez-Aviles L, Camacho L, Fernandez-Rodríguez C, et al. Hematopoietic clonal dominance, stem cell mutations, and evolutionary pattern of JAK2V617F allele burden in polycythemia vera. Eur. J. Haematol. 2015 Mar;94(3):251-7. doi.org/10.1111/ejh.12425.
- Barbui T, Thiele J, Carobbio A, Guglielmelli P, Rambaldi A, Vannucchi AM, et al. Discriminating between essential thrombocythemia and masked polycythemia vera in JAK2 mutated patients. Am. J Hematol. 2014 Jun;89(6):588-90. doi: 10.1002/ajh.23694.
- Roda P, Ferrari A, Tang X, Erlich P, Eisenhower C, Patel MD, et al. Determination of accuracy of polycythemia vera diagnoses and use of the JAK2V617F test in the diagnostic scheme. Ann Hematol. 2014 Sep;93(9):1467-72. doi: 10.1007/s00277-014-2068-2.
- Barbui T, Thiele J, Carobbio A, Gisslinger H, Finazzi G, Rumi E, et al. Masked polycythemia vera diagnosed according to WHO and BCSH classification. Am. J. Hematol. 2014 Feb;89(2):199-202. doi: 10.1002/ajh.23617.
- Ouyang J, Zheng W, Shen Q, Goswami M, Jorgensen JL, Medeiros LJ, et al. Flow cytometry immunophenotypic analysis of philadelphia-negative myeloproliferative neoplasms: correlation with histopathologic features. Cytometry. B Clin Cytom. 2014 Dec; 88(4):236-43. doi: 10.1002/cytob.21215.
- Ancochea AL, Morales IC, García PF, Martínez AL, Angona SA, Bellosillo B, et al. The role of serum erythropoietin level and jak2 v617f allele burden in the diagnosis of polycythaemia vera. Br J Haematol. 2014 Jul;167(3):411-7. doi: 10.1111/bjh.13047.
Correspondência
Danielle Cristinne Azevedo Feio
E-mail: [email protected]