The pathways of cell cycle regulation in retinoblastoma
As vias de regulação do ciclo celular em retinoblastoma
Renata Mendes de Freitas1
1Doutorado – Fundação Oswaldo Cruz – (Estágio PNPD) – Rio de Janeiro-RJ, Brasil.
Instituição: Fundação Oswaldo Cruz, Instituto Oswaldo Cruz, Rio de Janeiro. Laboratório de Epidemiologia de Malformações Congênitas Rio de Janeiro-RJ, Brasil.
Suporte financeiro: CAPES
Recebido em 27/05/2019
Artigo aprovado em 29/05/2019
DOI: 10.21877/2448-3877.201900853
INTRODUÇÃO
Retinoblastoma (RB) is an ocular tumor that affects retinal cells usually diagnosed in children up to 5 years of age. The prevalence is one in 15,000 – 20,000 born, being considered a rare neoplastic type, despite being the most frequent eye cancer in childhood.(1,2) Some scholars try to associate the higher incidence in developing countries, the level of education of the parents, the socioeconomic profile of the families, the professional qualification of the health care team and maternal nutritional conditions, emphasizing, for example, the amount of folic acid consumed during pregnancy.(3,4) The relationship between race, gender and ocular laterality in cases of retinoblastoma is not found in the studies conducted.(5)
This tumor type is associated with the biallelic loss of the RB1 tumor suppressor gene, which may be hereditary or sporadic, where the inhibition of retinoblastoma protein (pRB) function, encoded by the RB1 gene, is determinant in the genesis of retinoblastoma.(6) The loss of RB1 deregulates transcriptional factors associated with transcriptional activation of genes and triggers cellular apoptosis. In many types of cancers, loss of pRB can alter E2F and allow the deregulated maintenance of the cell cycle, but tumorigenesis can be prevented by p53-mediated surveillance.(7) In addition, in human tumors this event was also associated with the amplification of proteins encoded by the MDM2 and MDM4 genes, and could act as therapeutic targets for the treatment of retinoblastoma.(6)
Each component of this complex regulatory pathway pRB-E2F and associated genes in retinoblastoma will be addressed below.
The tumor suppressor gene RB1
In 1986, the RB1 gene was identified and cloned as the first tumor suppressor by Friend et al.(8) This gene, located in the chromosomal region 13q14, comprises more than 178,143 kb of genomic DNA and is composed of 27 exons that transcribe a messenger RNA of 4,772 kb. The complete gene sequence was reported by Toguchida et al. (1993).(9,10)
The RB1 gene encodes a nuclear protein of 928 amino acids called pRB, belonging to the pocket family, with ability to regulate gene transcription and act as regulator of the G1/S phases of the cell cycle.(11) pRB is also involved in the transcriptional regulation of genes that act on apoptosis, differentiation and cell adhesion.(12,13) The major cellular functions of pRB are conserved in mammalian cells, Drosophila melanogaster and Caenorhabditis elegans.(14)
Tracking for mutations in the RB1 gene should occur in all exons, with single base substitutions and large deletions being the most recurrent mutations. Many of these mutations are of the nonsense and frameshift type.(15) Most of the base substitutions originate from the transition from a cytosine to thymine (C> T) in the CGA codon into the amino acid arginine, generating a UGA stop codon, resulting in the loss of pRB function.(1,16)
Mutations detected in the RB1 gene may increase the quality of clinical management of retinoblastoma and allow predictions of risk for all family members.(17)
Complete depletion of the RB1 gene leads to increased chromosomal instability manifested in aneuploidy or polyploidy.(18) This phenomenon is attributed to centromeric dysfunction and failure to recruit components of the condensin II complex, which are proteins responsible for keeping both chromatid sisters together, leading to the defect of chromosome separation during mitosis.(18)
However, tumorigenesis occurs when surveillance pathways are inactivated, such as the p53 protein, that prevents the induction of apoptosis in the loss of pRB function.(19)
The retinoblastoma protein (pRB)
The RB protein (pRB) acts as a regulator of G1/S phases of the cell cycle and has the ability to regulate the gene transcription of genes that participate in apoptosis, differentiation and cell adhesion.(12,13)
This protein belongs to a family of proteins, encoded by three different genes: the RB1 gene, which encodes pRB; the RBL1 gene, located in the chromosomal region 20q11.2, which encodes p107, and the RBL2 gene, located on the long arm of chromosome 16, which encodes p130.(19,20) All members of the RB family of proteins have a conserved region, called pocket, which interacts with the LXCXE motif, found in viral proteins, used to bind to pRB. Deletions or point mutations in this domain make the oncoproteins incapable of inactivating pRB and potentiate cells in tumor targets.(21)
These proteins, known as “pocket”, are formed by three domains: N-terminal, A/B pocket and C-terminal.(20) Protein functionality is related to elongation factor 2 (E2F) binding domains.(10)
Cells expressing the mutant pRB, lacking the LXCXE interaction domain, showed abnormal chromatin structures, including decondensation of chromatin and loose chromosomes.(22) These data indicate that pRB also regulates chromatin remodeling, predominantly in the G0 and G1 phases of the cell cycle.(23)
The p107 and p130 proteins exert the function of regulating the repression of E2F target genes. However, such proteins are little mutated in human cancers.(13) Studies in mice showed that RB1 is an essential gene to the cells, and knockout mice die during the embryonic development. On the other hand, knockout mice, for the genes of the p107 and p130 proteins, usually develop suggesting that such proteins cannot replace all the functions of pRB and that this is in fact vital for the survival of the organism.(13)
In the quiescent phase of the cells (G0), the most abundant protein is p130; subsequently, cells are stimulated to initiate the cell cycle, and the pRB and p107 proteins are induced.(20) Thus, when the two begin to have their levels of expression increased, the levels of p130 are decreasing (Table 1). Some studies point to p130 as a partial transcriptional repressor called DREAM, and function to repress the functions of E2F target genes during the G0 phase.(20,24)
The initiation of gene transcription in eukaryotes requires that transcriptional protein factors, together with RNA polymerase, form a basal transcription apparatus to initiate gene transcription.(25)

The transcriptional factor in the control of the cell cycle:
The E2F transcription factor was initially identified as the cellular component required for the anterior region (E1A region) of the adenovirus protein, in order to mediate the transcriptional activation of the viral promoter region (E2 region).(26)
Chellappan et al. (1991) deduced the mechanism of regulation of E2F by activation of the adenovirus E1A region, allowing the understanding that the transcription factor is inhibited when associated with the RB protein. This observation was the first to suggest that E2F could be related to the development of cancer. (27) It was later identified that pRB is target of other viral oncoproteins, including SV40 of T antigen and human papillomavirus (HPV) E6 and E7 proteins.(28,29)
Subsequent studies have demonstrated that E2F controls the transcription of genes that are essential for cell division.(29)
This means that the HPV E7 viral protein is capable of binding to pRB and making it inactive; this way, the E2F factor becomes free, generating a stimulus for cell proliferation. In addition, the association of E7 protein with pRB leads to an increase in p53 that causes cell cycle arrest or induces apoptosis.(30) In the context of HPV infection, the E6 oncoprotein initiates the degradation of p53, leading to loss of control of cell proliferation and blockade in the activation of apoptosis.(30)
The ratio of pRB to cell cycle progression is associated with eight members of the E2F transcriptional factor family (E2F1 to E2F8) that are central regulators of cell cycle progression. The pRBs form heterodimers with a subunit encoded by DP – DP1 and DP2 family members, generating a DNA binding specificity, mainly determined by the E2F subunit.(12,13)
The factors E2F1, E2F2 and E2F3 are associated with transcriptional activation and have pRB as the target; E2F4 and E2F5 are transcriptional repressors and are targets of p107 and p130, whereas E2F6, E2F7 and E2F8 are independent repressors of pRB.(23) E2F1 plays an important role in the induction of apoptosis and is often found amplified in human cancers.(13)
Although the accepted model for the pRB-E2F complex reinforces that cyclin-dependent kinases (CDKs) phosphorylated pRB release the E2F transcription factors, during the G1/S cell cycle transition, it was observed that the pRB-E2F1 complexes persist associated beyond the S phase, independently of pRB phosphorylation. This complex will occupy promoter regions of apoptotic genes of proliferating cells.(31)
Thus, these observations indicate that phosphorylation of pRB by CDKs during the G1/S transition causes the release of most E2F factors to induce cell cycle gene transcription, but the pRB-E2F1 complexes remain stable thanks to the interaction of this complex to the promoter regions of apoptotic genes, repressing their expression.(31,32)
DeGregori (1997) suggests that E2F1-induced apoptosis is not simply a consequence of abnormality in S phase, but represents an intrinsic property of E2F1 in activating genes that initiate programmed cell death. It is possible that this ability of E2F1 causes direct or indirect interaction with other proteins participating in the apoptotic pathway, including p53. Thus, the results presented by him suggest the ability of E2F1 to induce an accumulation of p53, acting in the G1 phase of the cell cycle, promoting cell differentiation and control.(33)
Studies have suggested that E2F can bind to multiple promoter sites, allowing the ability to dynamically regulate the coding of genes that are important for DNA repair, chromatin remodeling, chromosomal segregation.(34)
The cell cycle activation by CDK
The proteins of the RB family are activated by phosphorylation of cyclin-dependent kinases (CDKs), which are divided into three classes – CDK2, CDK4 and CDK6. CDK2 associate with cyclins of type A and E; the CDK4 and CDK6 classes are associated with D-type cyclins (D1, D2 and D3) and are responsible for initiating pRB phosphorylation.(20) They act to maintain the cells in the G1 phase of the cell cycle, since they favor the maintenance of the pRB-E2F complex.(20) The CDK2/cyclin E complex promotes the progression of the G1 phase cycle to S phase, and CDK2/cyclin A is responsible for maintaining cell cycle progression after S phase.(6)
In its active form, pRB is hypophosphorylated in G0/G1 and associated with the transcriptional factor E2F, blocking the cell cycle. Extracellular factors that direct cell mitogenesis induce the activation of CDK2 and CDK4/CDK6-dependent cyclin-dependent kinases (CDKs) that promote phosphorylation of pRB.(28)
Once phosphorylated, the RB protein has its binding to E2F factor weakened and the dissociation of these molecules occurs, thereby allowing the onset and progression to the S-phase of the cell cycle.(29,35) The entire mechanism can be observed in Figure 1.
The inactivation of pRB promotes genomic instability and results in deletions, duplications and chromosomal rearrangements(35) and compromises the ability of cells to terminate the cell cycle, being highly susceptible to oncogenic gene proliferation.(12)
The literature shows that pRB is functionally impaired in many tumors as a result of RB1 mutations or mutations that increase phosphorylation of the protein or by the expression of viral oncoproteins targeting pRB.(16) Inactivation of pRB by oncogenic proteins is induced by viral infection and RB1 gene expression is suppressed via methylation of the DNA promoter region.(22)
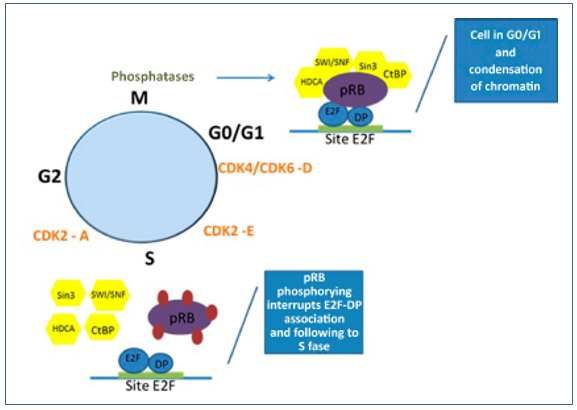
Figure 1. Mechanism of regulation of the pRB-E2F complex in cell cycle control. In G0/G1, the pRB-E2F complex is found with the active pRB bound to the transcriptional factor so that the cell cycle is halted in the G0/G1 phases and with highly condensed chromatin. With the activation of CDK4, CDK6 and CDK2-E in the G1 phase, the pRB-E2F complex is released and the cell cycle progresses through the S phase. The pRB is again activated by phosphatases, that allow the hypophosphorylation of the protein and stop the cell cycle in G0/G1.
With cell cycle progression, CDK activity is decreased by allowing the phosphatase 1 (PP1) protein to dephosphorylate pRB, promoting hypophosphorylation and allowing a new formation of the pRB-E2F complex, which represses gene transcription and cell cycle progression.(13)
This negative regulation of cell cycle progression is the main mechanism by which pRB represses tumor development. However, pRB interacts with hundreds of proteins that are critical to multiple processes beyond cell cycle control.(13)
Maintaining DNA integrity
The signaling pathway of the p53 protein, encoded by the TP53 gene, located on chromosome 17, is usually inactivated in several types of cancers; approximately 50% of all tumor types have mutations in the TP53 gene, and those with wild type p53 often show mutations in other signaling pathway genes that allow cell cycle surveillance.(36)
During a DNA damage response, the TP53 gene prevents cell proliferation through various mechanisms, such as cell cycle arrest and activation of apoptosis.(37)
In retinoblastoma, it is common for the TP53 gene not to be mutated, but other associated genes,(38,39) in which the deregulation of p53 stems from increased expression of regulatory genes such as MDM2 and MDM4.(40) The mdm2 and mdm4 proteins can prevent the effect of p53, playing a role of p53 regulators, inhibiting cell cycle control and preventing apoptosis.(37)
Increased expression of MDM2 and MDM4 genes can be activated by somatic amplifications in tumors or germ polymorphisms, leading to a predisposition of individuals to cancer.(40)
In addition to the increased expression of the p53 signaling pathway antagonist genes, there are several splicing variants in the formation of messenger RNAs of the MDM2 and MDM4 genes leading to p53 repression.(36)
CDKIs activation and cell cycle arrest
The mechanism of activation of CDKs can be regulated by inhibitory proteins known as CDKI – cyclin-independent kinase inhibitor, mainly acting in the control of the G1 and S phases of the cell cycle. There are two classes of CDKI: the CIP family and KIP, which consists of the p21, p27, p57, p18 and p19 proteins. These members bind cyclin and CDK subunits, inhibiting kinase activity (Figure 2).(18)
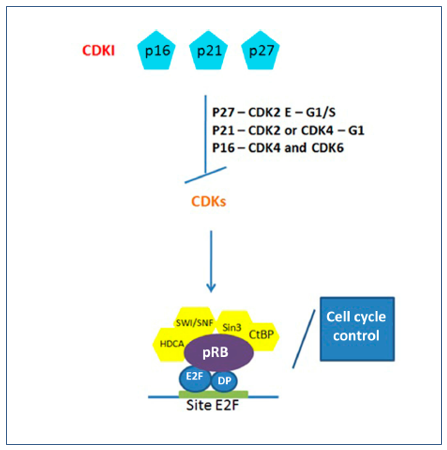
Figure 2. Regulation of CDKs by action of CDKIs and restoration of pRB-E2F complex in cell cycle control. The main CDKIs in the control of CDKs in the pathogenic pathway of retinoblastoma and cell cycle arrest due to hypophosphorylation of pRB and formation of the pRB-E2F complex.
The CDKN1B gene, located in the chromosomal region 12p13, encodes the p27 kinase inhibitor, which inhibits the CDK2/E complex, negatively regulating cell cycle progression in the G1/S phases and promoting cycle arrest in G1 phase.(18) In this way, p27 controls proliferation, differentiation, cell adhesion and apoptosis. In G1 the expression levels of p27, for example, are relatively high and CDK2/E activity is low, preventing DNA synthesis.(18)
The low expression of CDKN1B is related to tumorigenesis and the advancement of the clinical stage in several tumors, such as prostate, gastric, laryngeal, colorectal and breast. Some SNPs (single nucleotide polymorphism) in the gene are associated with reduced transcription rate and low levels of p27.(41) Such SNPs may affect the normal functioning of CDKN1B by altering the efficiency of transcription and protein levels, leading to cell cycle deregulation and neoplastic behavior.(41)
Some studies have shown increased susceptibility to cancer with the identification of polymorphism rs2066827.(41) Landa et al. (2010) (42) suggests that this polymorphism (T326G, V109G, rs2066827) in the CDKN1B gene is associated with reduced transcription rate and low protein levels with susceptibility to thyroid and prostate cancer.(42)
P27 was identified in the association of multiple endocrine neoplasm syndrome type 1 (MEN-1) with mutations of loss of germline function in the CDKN1B gene in mice. Expression studies for p27 showed an absence or reduced presence in normal and pathological tissues of these mice.(43) In vitro cell culture studies have demonstrated that the mutant p27 protein retains some properties of the wild-type protein, such as nuclear localization and interaction with CDK2/E, but is highly unstable and rapidly degraded.(43)
The key role of the CDKN1B gene in the regulation of cell cycle proliferation and as a tumor suppressor has been demonstrated in knockout mice that showed gigantism due to increased cell number and increased tumorigenesis.(44)
Another CDK control gene is CDKN1A, located in the chromosomal region 6p21.2, which encodes the cyclin-dependent inhibitory kinase (CDKI) p21, which associates with the CDK2 or CDK4 complexes, inhibiting the activity of these complexes and regulating the progression of the cycle cells in G1.(23) In addition, p21 protein may also play a regulatory role in S phase during cell duplication and DNA repair.(45) This signaling control is mediated by p53, which induces p21 expression in response to DNA damage, disrupting cell cycle progression in the transition from G1 phase to S phase by inhibition of CDKs.(45)
Sheikh et al. (1994)(46) have demonstrated in a study of breast cancer cells that mechanisms regulating p21 expression involve p53-dependent and p53-independent signaling pathways. In ovarian cancer cells, it has been observed that the induction of p21 expression can occur by activation of protein kinase C (PKC) in cells lacking p53.(47)
A study (2001) demonstrated that p21 expression is associated with the presence of metastases and probably with great genetic instability.(48)
The CDKN2A gene, located in the chromosomal region 9p21.3, generates variant transcripts that differ in the first exon and at least three alternatively processed variants encoding different proteins have been described, two of which encode structurally related isoforms that function as inhibitors of CDK4 kinase. The remaining transcript contains an open reading frame (denominated ARF) which specifies a protein that is structurally unrelated to the products of the other variants. This ARF product functions as a stabilizer of the p53 protein, since it can interact and inhibit mdm2, a protein responsible for the degradation of p53 (Figure 3).(49)
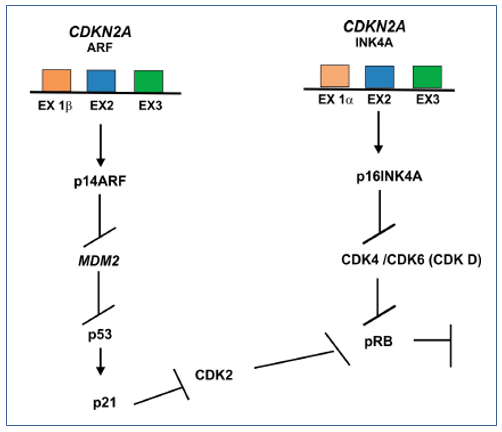
Figure 3. p14ARF inhibiting mdm2 also blocks the activity of p53 but activates p21 that will inhibit CDK2 and block pRB. A p16 is a CDK inhibitor that limits the activity of CDK4 and CDK6 complexes, disrupting their interaction with D-type cyclins. Thus, p16 regulates the phosphorylation of pRB by inhibiting CDK4 or by releasing specific CDK6 inhibitors, resulting in non phosphorylated pRB, keeping the cells in the G1 phase.(1,26)
Despite the structural and functional differences between the CDK inhibitory isoforms and the ARF product encoded by this gene, through the regulatory roles of CDK4 and p53 in cell cycle G1 progression, they share a common functionality in the control of G1. This gene is often mutated or deleted in a wide variety of tumors, and is known to be a major tumor suppressor gene.(49) The activating signals of p16 are diverse, including those that promote cell cycle arrest due to the presence of DNA damage or to those associated with tumorigenesis.(50)
Regulators genes of the p53 activation pathway
The MDM2 gene, mapped in the 12q15 chromosomal region, is the major regulator of p53, keeping the protein at appropriate levels in normal cells. After stress, p53 is activated and induces apoptosis.(51)
The mdm2 acts as a ubiquitin ligase that binds to p53 for its destruction via proteosomes, preventing apoptosis from occurring in cells. Cells with damaged DNA activate protein kinases, which phosphorylate p53 and reduce binding with mdm2 allowing apoptosis.(31,39) Thus, an increase in mdm2 levels has been observed in a broad spectrum of human cancers.(45,52)
The expression of mdm2 has been shown in previous studies, resulting in a decrease in E2F1 levels, causing mdm2 to inhibit the apoptosis promoted by the transcriptional factor. In this way, mdm2 is a negative regulator of E2F1 and also of p53.(52,20)
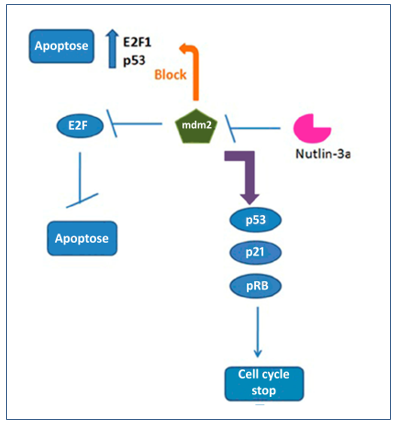
Figure 4. Regulation of Nutlin-3a in mdm2, E2F, p53, p21, pRB, apoptosis and cell cycle arrest. Nutlin-3a when it blocks mdm2, also blocks E2F and consequently apoptosis. Thus, with the blockade of mdm2, the accumulation of p53, p21, pRB occurs, restoring the formation of the pRB-E2F complex, stopping the cell cycle and enabling the activation of apoptosis.
Nutlin-3a consists of an inhibitor of mdm2. A recent study showed that this substance causes p53 and p21 accumulation and pRB hypophosphorylation, which led to cell cycle arrest in some cell lines.(20) On the other hand, in other strains, nutlin-3a downregulated pRB and resulted in apoptosis independent of E2F1. Thus, nutlin-3a is considered a potential therapeutic agent that could suppress and/or kill tumor cells. However, the mechanism by which this substance acts by inducing hypophosphorylation of pRB in some cells is not well understood.(20)
Another gene, MDM4, located at 1q32, regulates the stability of p53 in a process dependent on mdm2. The mdm4 protein, belonging to the same mdm2 family, shares several structural and functional properties, including a highly conserved p53 binding domain capable of inhibiting p53 transactivation.(52)
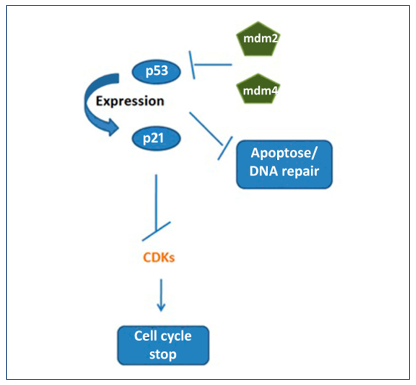
Figure 5. Normally, p53 activates p21 expression leading to inactivation of CDKs and cell cycle arrest. The mdm2 and mdm4 proteins when blocking p53 also block apoptosis and DNA repair.
In addition to binding to p53 and blocking apoptosis, mdm4 has the ability to increase the stability of p53 by binding to mdm2 through a conserved domain so that the interaction between mdm2-mdm4 complexes inhibits targeted p53 degradation by mdm2.(52)
The mdm4 protein also regulates pRB levels by mdm2-mediated ubiquitination, being inhibited by mdm4 due to competition between mdm2-mdm4 by binding sites in the C-terminal domain of pRB.(39) Polymorphisms and mutations in MDM4 have been frequently studied because of the impact on the regulation of p53 and pRB and the association of risk for some types of cancer.(53)
CONCLUSION
In most cancers, these pathways are impaired by mutations in TP53, inactivation of the CDKN2A gene or amplification of MDM2. Nutlin-3a-like molecules weaken the mdm2-p53 interaction by impairing the survival of tumor cells in retinoblastoma.(54) Expression of MDM4 in the absence of MDM2 suggested a mode of regulation of p53. With the absence of MDM2, the effects of Nutlin-3a were related to inhibition of tumorigenesis mainly by blocking the mdm4-p53 interaction.(54) Previous studies have shown that the high level of MDM2 expression is crucial for the proliferation and survival of retinoblastoma cells.(54)
Many functions of pRB are related to genomic instability, cancer associated with poor prognosis, tumor heterogeneity and development of therapeutic resistance. Signaling pathways that influence pRB and retinoblastoma have been shown here. All components are critical for understanding cell cycle control and activation of apoptosis. Loss-of-function mutations in each molecule comprising this large pathway can generate responses that alter cellular behavior, leading to uncontrolled cell proliferation, chromosomal instability and tumorigenesis.(54)
Abstract
Retinoblastoma is a childhood ocular tumor often caused by the biallelic inactivation of the RB1 gene affecting children up to 5 years of age. A retinoblastoma protein (pRB), encoded by the tumor suppressor gene RB1, is responsible for the regular progression of the G1 phase to the phase S of the cell cycle. This protein forms a complex with the transcriptional factor E2F causing the cell cycle to remain in the G0/G1 stage. With a phosphorylation of cyclin-dependent kinases (CDK), the phosphorylation of the RB protein is activated and the complex formed with E2F is disrupted, with the advancement of the cell cycle to an S phase and cell proliferation. All the control of cell proliferation is regulated not only by the complex formed by RB and E2F proteins, but also by other proteins that participate in and/or interfere in this cell division control mechanism, such as mdm2, mdm4 and p21 proteins.
Keywords
Retinoblastoma protein; retinoblastoma; cell cycle proteins
REFERENCES
- Mohd Khalid MK, Yakob Y, Md Yasin R, Wee Teik K, Siew CG, Rahmat J, et al. Spectrum of germ-life RB1 gene mutations in Malaysian patients with retinoblastoma. Mol Vis. 2015; 21:1185-90.
- Scollon S, Anglin AK, Thomas M, Turner JT, Wolfe Schneider K. A comprehensive review of pediatric tumors and associated cancer predisposition syndromes. J Genet Couns. 2017;26(3):387-43.
- Orjuela MA, Cabrera-Muñoz L, Paul L, Ramirez-Ortiz MA, Liu X, Chen J, et al. Risk of retinoblastoma is associated with a maternal polymorphism in dihydrofolate reductase (DHFR) and prenatal folic acid intake. Cancer. 2012 Dec 1;118(23):5912-9.
- Rodriguez-Galindo C, Orbach DB, VanderVeen D. Retinoblastoma. Pediatr Clin North Am. 2015 Feb;62(1):201-23.
- Selistre SGA, Maestri MK, Santos-Silva P, Schüler-Faccini L, Guimarães LSP, Giacomazzi J, et al. Retinoblastoma in a pediatric oncology reference center in Southern Brazil. BMC Pediatr. 2016;16:48.
- Knudsen ES, Knudsen KE. Tailoring to RB: tumor suppressor status and therapeutic response. Nat Rev Cancer. 2008;8(9):714-24
- Laurie NA, Donovan SL, Shih CS, Zhang J, Mills N, Fuller C, et al. Inactivation of the p53 pathway in retinoblastoma. Nature. 2006 Nov 2;444(7115):61-6.
- Benavante CA, Dyer MA. Genetics and epigenetics of human retinoblastoma. Annu Rev Pathol. 2015;10:547-62
- Toguchida J, McGee TL, Paterson JC, Eagle JR, Tucker S, Yandell DW et al. Complete genomic sequence of the human retinoblastoma susceptibility gene. Genomics. 1993;17(3):535-43.
- Kalsoom S, Wasim M, Afzal S, Shahzad MS, Ramzan S, Awan AR, et al. Alterations in the RB1 gene in Pakistani patients with retinoblastoma using direct sequencing analysis. Mol Vis. 2015 Sep 17;21:1085-92.
- Dyson N. The regulation of E2F by pRB- family proteins. Genes Dev. 1998;12(15):2245-62.
- Dyson NJ. RB1: a prototype tumor suppressor and an enigma. Genes Dev. 2016;30(13):1492-502.
- Vélez-Cruz R, Johnson DG. The retinoblastoma (RB) tumor suppressor: pushing back against genome instability on multiple fronts. Int J Mol Sci. 2017 Aug 16;18(8). pii: E1776).
- Sage J. The retinoblastoma tumor suppressor and stem cell biology. Genes Dev. 2012 Jul 1;26(13):1409-20.
- Ottaviani D, Alonso C, Szijan I. Uncommon RB1 somatic mutations in a unilateral retinoblastoma patient. Medicina (B Aires). 2015;75 (3):137-41.
- Mateu E, Sánchez F, Nájera C, Beneyto M, Castell V, Hernández M, et al. Genetics of retinoblastoma: a study. Cancer Genet Cytogenet.1997;95(1):40-50.
- Ahani A, Akbari MT, Saliminejad K, Behnam B, Akhondi MM, Vosoogh P, et al. Screening for large rearrangements of the RB1 gene in Iranian patients with retinoblastoma using multiplex ligation-dependent probe amplification. Mol Vision. 2013;19:454-62.
- Huang PH, Cook R, Mittnacht S. RB in DNA repair. Oncotarget. 2015;6(25):20746-7.
- Di Fiore R, D’Anneo A, Tesoriere G, Vento R. RB1 in cancer: different mechanisms of RB1 inactivation and alterations of pRb pathway in tumorigenesis. J Cell Physiol. 2013 Aug;228(8):1676-87.
- Henley SA, Dick FA. The retinoblastoma family of proteins and their regulatory functions in the mammalian cell division cycle. Cell Div. 2012 Mar 14;7(1):10.
- Dahiya A, Gavin MR, Luo RX, Dean DC. Role of the LXCXE binding site in Rb function. Mol Cell Biol. 2000;20(18):6799-805.
- Uchida C. Roles of pRB in the regulation of nucleosome and chromatin structures. Biomed Res Int. 2016;2016:5959721.
- Longworth MS, Dyson NJ. pRB, a local chromatin organizer with global possibilities. Chromosoma. 2010 Feb;119(1):1-11.
- Engeland K. Cell cycle arrest through indirect transcriptional repression by p53: I have a DREAM. Cell Death Differ. 2018 Jan; 25(1):114-132.
- Pimentel M, Rebouças CS, Gallo C. Genética Essencial. Rio de Janeiro: Editora Guanabara Koogan; 2013.
- Yee AS, Reichel R, Kovesdi I, Nevins JR. Promoter interaction of the E1A-inducible factor E2F and its potential role in the formation of a multicomponente complex. EMBO J. 1987 Jul;6(7):2061-8.
- Chellappan SP, Hiebert S, Mudryj M, Horowitz JM, Nevins Jr. The E2F transcription factor is a cellular target for the RB protein. Cell. 1991 Jun 14;65(6):1053-61.
- Sellers WR, Kaelin WG Jr. Role of the retinoblastoma protein in the pathogenesis of Human Cancer. J Clin Oncol. 1997;15(11):3301-12.
- Bell LA, Ryan KM. Life and death decisions by E2F-1. Cell Death Differ. 2004;11(2):137-42.
- Fischer M, Uxa S, Stanko C, Magin TM, Engeland K. Human papilloma virus E7 oncoprotein abrogates the p53-p21-DREAM pathway. Sci Rep. 2017 Jun 1;7(1):2603.
- Indovina P, Pentimalli F, Casini N, Vocca I, Giordano A. RB1 dual role in proliferation and apoptosis: Cell fate control and implications for cancer therapy. Oncotarget. 2015 Jul 20;6(20):17873-90.
- Dick FA, Rubin SM. Molecular mechanisms underlying RB protein function. Nat Rev Mol Cell Biol. 2013;14(5):297-306.
- Perri F, Pisconti S, Della Vittoria Scarpati G. P53 mutations and cancer: a tight linkage. Ann Transl Med. 2016;4(24):522.
- Crosby ME, Almasan A. Opposing roles of E2Fs in cell proliferation and death. Cancer Biol Ther. 2004;3(12):1208- 11.
- Ganguly A, Shields CL. Differential gene expression profile of retinoblastoma compared to normal retina. Mol Vis. 2010;16:1292-30.
- McEvoy JD, Dyer MA. Genetic and epigenetic discoveries in human retinoblastoma. Crit Rev Oncog. 2015; 20(3-4): 217-25.
- Jiao Y, Jiang Z, Wu Y, Chen X, Xiao X, Yu H. A functional polymorphism (rs937283) in the MDM2 promoter region is associated with poor prognosis of retinoblastoma in Chinese Han population. Sci Rep. 2016 Aug 10;6:31240.
- Kato MV, Shimizu T, Ishizaki K, Kaneko A, Yandell DW, Toguchida J, et al. Loss of heterozygosity on chromosome 17 and mutation of the p53 gene in retinoblastoma. Cancer Lett. 1996 Aug 23;106(1):75-82.
- McEvoy J, Ulyanov A, Brennan R, Wu G, Pounds S, Zhang J, et al. Analysis of MDM2 and MDM4 single nucleotide polymorphisms, mRNA splicing and protein expression in retinoblastoma. Plos One. 2012; 7(8), e42739.
- de Oliveira Reis AH, de Carvalho IN, de Sousa Damasceno PB, Ferman SE, Lucena E, Lopez-Camelo JS, et al. Influence of MDM2 and MDM4 on development and survival in hereditary retinoblastoma. Pediatr Blood Cancer. 2012 Jul 15;59(1):39-43.
- Lu Y, Gao K, Zhang M, Zhou A, Zhou X, Guan Z, et al. Genetic association between CDKN1B rs2066827 polymorphism and susceptibility to cancer. Medicine (Baltimore). 2015;94(46): e1217.
- Landa I, Montero-Conde C, Malanga D, De Gisi S, Pita G, Leandro-Garcia LJ, et al. Allelic variant at -79(C>T) in CDKN1B (p27Kip1) confers an increased risk of thyroid cancer and alters mRNA levels. Endocr Relat Cancer. 2010 Jun 1;17(2):317-28.
- Tonelli F, Giudici F, Giusti F, Marini F, Cianferotti L, Nesi G, et al. A heterozygous frameshift mutation in exon 1 of CDKN1B gene in a patient affected by MEN4 syndrome. Eur J Endocrinol. 2014;171 (2):K7-K17.
- Chang BL, Zheng SL, Isaacs SD, Wiley KE, Turner A, Li Ge, et al. A polymorphism in the CDKN1B gene is associated with increased risk of hereditary prostate cancer. Cancer Res. 2004;64 (6):1997-9.
- Chen R, Liu S, Ye h, Li J, Du Y, Chen L, et al. Association of p53 rs1042522, MDM2 rs2279744, and p21 rs1801270 polymorphisms with retinoblastoma risk and invasion in a Chinese population. Sci Rep. 2015 Aug 20;5:13300
- Sheikh MS, Li XS, Chen JC, Shao ZM, Ordonez JV, Fontana JA. Mechanisms of regulation of WAF1/Cip1 gene expression in human breast carcinoma: role of p53-dependent and independent signal transduction pathways. Oncogene. 1994;9(12):3407-15.
- Akashi M, Osawa Y, Koeffler HP, Hachiya M.. p21WAF1 expression by an activator of protein kinase C is regulated mainly at the post-transcriptional level in cells lacking p53: important role of RNA stabilization. Biochem J. 1999 Feb 1;337( Pt 3):607-16.
- Ceccarelli C, Santini D, Chieco P, Lanciotti C, Taffurelli M, Paladini G, et al. Quantitative p21waf1/p53 immunohistochemical analysis defines groups of primary invasive breast carcinomas with different prognostic indicators. Int J Cancer. 2001 Mar 20;95(2):128-34.
- Sharpless NE, Sherr CJ. Forging a signature of in vivo senescence. Nat Rev Cancer. 2015 Jul;15(7):397-408. Erratum in: Nat Rev Cancer. 2015 Aug;15(8):509.
- Gonzalez-Gomez P, Bello MJ, Alonso ME, Arjona D, Lomas J, de Campos JM, et al. CpG island. Methylation status and mutation analysis of the RB1 gene essential promoter region and protein-binding pocket domain in nervous system tumors. Br J Cancer. 2003 Jan 13;88(1):109-14.
- Aberg E, Saccoccia F, Grabherr M, Ore WYJ, Jemth P, Hultqvist G. Evolution of the p53-MDM2 pathway. BMC Evol Biol. 2017 Aug 3;17(1):177.
- Strachan GD, Rallapalli R, Pucci B, Lafond TP, Hall DJ. A transcriptionally inactive E2F-1 targets the MDM family of proteins for proteolytic degradation. J Biol Chem. 2001 Dec 7;276(49):45677-85.
- Reincke S, Govbakh L, Wilhelm B, Jin H, Bogdanova N, Bremer M, et al. Mutation analysis of the MDM4 gene in German breast cancer patients. BMC Cancer. 2008 Feb 15;8:52.
- Qi DL, Cobrinik D. MDM2 but not MDM4 promotes retinoblastoma cell proliferation through p53-independent regulation of MYCN translation. Oncogene. 2017 Mar 30;36(13):1760-1769.
Correspondência
Renata Mendes de Freitas
Av. Brasil, 4353, Manguinhos
Pavilhão Leônidas Deane, 6º andar, sala 617
21040-900 – Rio de Janeiro-RJ, Brasil